스플라이스 변이를 통한 인구 적합도와 유전 부하 분석
초록
본 연구는 정보 이론 기반의 개별 정보(Ri) 변화를 이용해 mRNA 스플라이스 부위에 영향을 미치는 SNP들의 적합도와 유전 부하를 정량화한다. HapMap 데이터의 4백만 개 이상의 SNP를 분석해 스플라이스 부위의 정보 손실·증가를 계산하고, 이를 대립유전자 빈도와 결합해 유전 부하(L)를 추정하였다. 높은 부하(L > 0.5)를 가진 SNP가 흔히 존재함을 확인했으며, 일부는 마이크로어레이 기반 발현 데이터와 일치해 실제 스플라이스 교란을 시사한다.
상세 분석
이 논문은 전통적인 GWAS가 제공하는 연관성 정보만으로는 기능적 위험 대립유전자를 정확히 식별하기 어렵다는 점을 지적하고, 정보 이론(Shannon entropy)을 활용한 새로운 정량적 접근법을 제시한다. 핵심은 스플라이스 부위의 결합 친화도를 비트 단위의 개별 정보(Ri)로 표현하고, 변이 전후의 ΔRi를 측정함으로써 변이가 결합 효율에 미치는 영향을 직접적으로 추정한다는 점이다. 저자는 Ri와 복제율(W) 사이에 로그 관계(Ri ≈ log₂W)를 가정하고, 이를 바탕으로 두 대립유전자의 적합도 차이를 ΔRi로 환산한다. 이후 각 대립유전자의 빈도(p)와 적합도(w)를 가중 평균해 평균 적합도(Ȳ)를 구하고, 최대 가능한 적합도(w_max)와의 차이를 이용해 유전 부하 L = 1 − Ȳ/w_max 를 정의한다. 이 수식은 다중 유전자 좌위가 독립적이라고 가정하므로, 실제 연관성(링키지 디시퀸스)나 상호작용을 무시한다는 제한점이 있다.
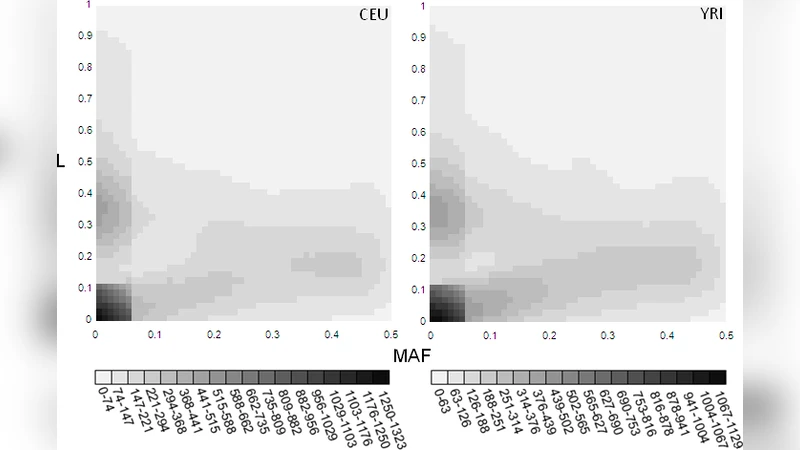
방법론에서는 인간 전체 유전체(hg18)와 HapMap Phase II SNP 데이터를 결합해, 알려진 donor·acceptor 스플라이스 서열에 대한 정보 가중 행렬을 적용하였다. 평균적으로 donor는 9.8118 bit/site, acceptor는 8.1214 bit/site의 정보를 갖는다. 모든 SNP에 대해 ΔRi를 계산하고, 변이가 자연 스플라이스 부위에 미치는 영향을 정량화하였다. 변이된 부위가 cryptic site를 활성화하거나 기존 site의 친화도를 감소시키는 경우를 구분하고, ΔRi가 0.1 bit 이상인 경우를 ‘유의미한 변화’로 간주하였다.
결과적으로 4,071,589개의 SNP 중 1,093,474개가 스플라이스 부위의 정보를 변화시켰으며, 그 중 9,051개가 실제 유전자의 donor·acceptor 부위에 영향을 주었다. ΔRi가 0.5 bit 이상인 SNP는 전체의 약 70 %를 차지했으며, 이들 중 다수는 높은 유전 부하(L > 0.5)를 보였다. 흥미롭게도 높은 부하를 가진 SNP가 흔히 높은 대립유전자 빈도(MAF > 0.05)를 갖는 경우가 관찰되었는데, 이는 선택압이 완전히 제거되지 않은 ‘중립적’ 변이일 가능성을 시사한다. 또한, 변이된 부위의 ΔRi와 마이크로어레이 기반 스플라이스 인덱스(SI) 간의 상관관계를 검증했으나, ΔRi가 0.5 bit 이하인 경우 SI 변화가 미미하거나 통계적으로 유의하지 않은 경우가 많았다. 이는 작은 정보 손실이 실제 전사 수준에서 감지되기 어려움을 반영한다.
한계점으로는 (1) 스플라이스 부위 외의 조절 요소(예: 스플라이스 인핸서, RNA‑binding protein)와의 상호작용을 고려하지 않았으며, (2) 복제율을 로그 형태로 단순화함으로써 실제 생물학적 복잡성을 축소했다는 점이다. 또한, 유전 부하 계산이 다중 좌위 간 독립성을 전제로 하므로, LD 블록 내에서의 누적 효과는 과소평가될 수 있다. 그럼에도 불구하고, 정보 이론 기반의 정량적 프레임워크는 SNP 기능 예측과 인구 수준의 적합도 평가를 연결하는 유용한 도구임을 보여준다.
댓글 및 학술 토론
Loading comments...
의견 남기기