GWAS 데이터로 복합질환 CNV 탐색 전략
초록
본 논문은 인간 유전체에서 복제수 변이(CNV)가 SNP보다 더 많은 변이를 차지한다는 점을 강조하고, GWAS 플랫폼에서 얻은 하이브리다이제이션 강도와 형질점수를 이용해 CNV를 검출·분석하는 최신 방법들을 정리한다. 또한 흔한 CNV와 희귀 CNV를 구분해 각각에 적합한 검출 전략과 통계 모델을 제시하며, 변이의 질병 기여도를 평가하기 위한 실용적인 권고사항을 제공한다.
상세 분석
논문은 먼저 인간 유전체에서 CNV가 차지하는 비중과 구조적 특성을 정량화한다. CNV는 수천 베이스쌍에서 수백 메가베이스까지 다양하며, 유전자와 조절 영역을 모두 포함하므로 기능적 효과가 클 가능성이 있다. 이러한 배경에서 GWAS에서 사용되는 SNP 마이크로어레이와 최신 옥시포드(oxford) 플랫폼은 각 프로브의 형광 강도(intensity)와 알레일 비율(B‑allele frequency, BAF)를 동시에 제공한다는 점을 활용한다. 형광 강도는 복제수에 비례하고, BAF는 이형접합 비율을 반영하므로 두 신호를 결합하면 복제수의 0,1,2,3… 등 다양한 상태를 추정할 수 있다.
통계적 모델로는 Hidden Markov Model(HMM), Gaussian Mixture Model(GMM), 그리고 최근에는 베이지안 네트워크와 딥러닝 기반 변이 검출기가 소개된다. HMM은 연속적인 프로브 신호를 상태 전이 확률로 모델링해 CNV 구간을 연속적으로 추정하는 데 강점이 있다. GMM은 복제수별로 정규분포를 가정하고, EM 알고리즘을 통해 파라미터를 최적화한다. 두 방법 모두 잡음이 큰 데이터에서 과잉 검출을 방지하기 위해 사전 확률과 최소 길이 제한을 도입한다.
흔한 CNV(MAF > 5%)를 탐색할 때는 ‘태그 SNP’ 접근법이 유용하다. 특정 CNV와 강하게 연관된 SNP를 식별해, 해당 SNP를 대리 마커로 사용하면 비용과 계산량을 크게 절감할 수 있다. 그러나 태그 SNP는 복제수 변이의 복잡성을 완전히 포착하지 못하고, 특히 복제수 증가(duplication)와 감소(deletion)를 구분하기 어렵다. 따라서 형광 강도와 BAF를 직접 이용하는 방법이 더 높은 검출력과 정확도를 제공한다는 것이 논문의 핵심 결론이다.
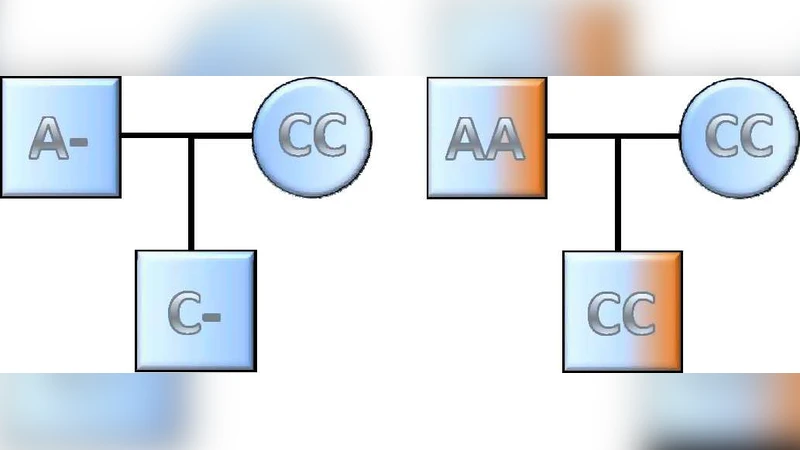
희귀 CNV(MAF < 1%)는 케이스‑컨트롤 설계에서 통계적 파워가 급격히 떨어진다. 이를 보완하기 위해 ‘de novo’ CNV 탐색이 강조된다. 부모‑자식 삼인조(trio) 데이터를 이용해 자식에게서만 나타나는 새로운 CNV를 식별하면, 질병 연관성을 높은 신뢰도로 추정할 수 있다. 또한, 고해상도 시퀀싱과 결합한 ‘read depth’ 분석, ‘split‑read’ 및 ‘discordant pair’ 신호를 활용하면 초소형 CNV도 검출 가능하다.
마지막으로 논문은 데이터 전처리(배경 보정, 프로브 필터링), 품질 관리(QC) 지표(예: LRR 표준편차, BAF 드리프트), 그리고 다중 검정 보정 방법을 상세히 제시한다. 연구자는 CNV 검출 파이프라인을 구축할 때, 여러 알고리즘을 병합해 합의(consensus) 결과를 도출하고, 독립적인 검증 코호트를 활용해 재현성을 확인할 것을 권고한다.
댓글 및 학술 토론
Loading comments...
의견 남기기