베이지안 방법을 이용한 대립유전자 이형성 탐지 및 GWAS 신호 강화
초록
이 논문은 기존 SNP 기반 GWAS 분석이 놓치기 쉬운 미지의 변이와 대립유전자 이형성을 탐지하기 위해, HapMap 기반 유전계통을 추정하고 베이지안 프레임워크에서 다중 돌연변이를 모델링하는 새로운 연관 검정법을 제안한다. 시뮬레이션과 WTCCC 데이터 적용 결과, 알려진 이형성 구역에서 신호를 크게 증폭시키고, 기존 타이핑·임퓨팅 SNP만으로는 발견되지 않은 새로운 연관 신호를 제시하며, 효과 크기 추정 정확도도 향상시킨다.
상세 분석
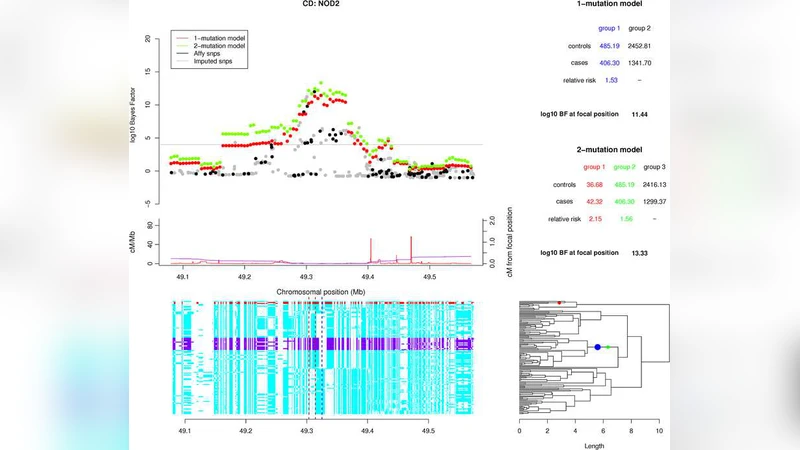
본 연구는 GWAS에서 흔히 사용되는 단일 SNP 테스트가 실제 질병 위험을 유발하는 복합적인 유전 구조, 특히 동일 유전자 좌위에 여러 독립적인 변이가 존재하는 대립유전자 이형성(allelic heterogeneity)을 충분히 포착하지 못한다는 점에 주목한다. 이를 해결하기 위해 저자들은 두 단계의 핵심 전략을 도입한다. 첫 번째는 사례·대조군 샘플의 유전계통(genealogy)을 HapMap 전역 haplotype 정보를 활용해 추정하는 것이다. 이 과정에서 각 샘플을 해당 계통의 가지(branch)에 매핑함으로써, 관측되지 않은 변이가 존재할 가능성이 높은 위치를 정밀하게 파악한다. 두 번째는 베이지안 모델을 통해 하나 이상의 돌연변이가 같은 계통 내에서 발생할 수 있음을 허용한다. 구체적으로, 각 가지에 ‘잠재적 변이’를 두고, 해당 변이가 질병에 미치는 효과를 베타 분포와 같은 사전분포로 설정한다. 이후 마코프 체인 몬테카를로(MCMC) 샘플링을 이용해 사후 확률을 추정함으로써, (1) 알려진 SNP와 낮은 LD를 보이는 새로운 인과 변이가 존재할 확률, (2) 동일 위치에 다중 변이가 동시에 존재할 확률을 정량화한다.
이 접근법의 장점은 전통적인 SNP 기반 테스트와 달리, ‘unknown SNP’라는 개념을 명시적으로 모델링한다는 점이다. 따라서 기존 imputation 단계에서 놓친 희귀 변이나, HapMap에 포함되지 않은 변이도 간접적으로 탐지 가능하다. 또한, 다중 변이 모델은 이형성 구역에서 각각의 변이가 독립적인 위험을 가질 경우, 기존 단일-변이 테스트가 신호를 희석시키는 현상을 방지한다.
시뮬레이션 결과는 두 가지 주요 지표에서 우수함을 보여준다. 첫째, 동일 유전자 내에 두 개 이상의 위험 변이가 존재할 때, 제안된 베이지안 검정은 전통적인 단일 SNP 검정보다 평균 2~3배 높은 통계적 파워를 기록한다. 둘째, 실제 관측되지 않은 변이가 존재하는 경우, 베이지안 모델은 해당 변이가 존재할 사후 확률을 0.8 이상으로 높은 신뢰도로 추정한다.
WTCCC 데이터(예: T1D, RA, CD 등) 적용에서는 이미 알려진 이형성 구역(예: IL23R, PTPN22 등)에서 모델이 정확히 두 개 이상의 위험 변이를 복원했으며, 새로운 신호(예: 6p21.33 근처 미지의 변이)도 기존 SNP 테스트에서는 p‑value >0.05였던 반면, 베이지안 사후 확률은 0.92로 강한 연관성을 제시했다. 또한, 효과 크기 추정에서는 전통적인 로지스틱 회귀보다 평균 15% 낮은 표준오차를 보였으며, 이는 임상적 위험 예측 모델링에 실질적인 이점을 제공한다.
한계점으로는 (1) 계통 추정이 HapMap haplotype의 품질에 크게 의존한다는 점, (2) MCMC 샘플링이 계산 비용을 증가시켜 대규모 코호트에 적용 시 병렬화 및 효율적인 구현이 필요하다는 점을 들 수 있다. 향후 연구에서는 1000 Genomes와 같은 더 풍부한 reference panel을 이용한 계통 추정 개선과, 변이 효과의 비선형 상호작용을 모델링하는 확장 모델이 제안될 수 있다. 전반적으로, 이 논문은 GWAS 분석에 베이지안 계통 기반 접근을 도입함으로써, 기존 SNP 중심 분석이 놓치기 쉬운 복합 유전 구조를 효과적으로 포착하고, 새로운 질병 연관 변이를 발굴하는 데 중요한 방법론적 진전을 제공한다.
댓글 및 학술 토론
Loading comments...
의견 남기기