제약 논리 프로그래밍 기반 단백질 조각 조립
초록
본 논문은 제약 논리 프로그래밍(CLP)을 이용해 단백질 구조 예측을 수행한다. 알려진 단백질 데이터베이스에서 유사도와 출현 빈도에 따라 조각을 추출·클러스터링하고, 이를 중간 정밀도의 Ca‑측쇄 중심 모델에 매핑한다. 조각 배치를 제약 만족 문제로 정의하고, 에너지 함수와 대규모 이웃 탐색을 결합해 최적 구조를 찾는다. 실험 결과, 제안 방법이 효율적이며 향후 다양한 조각 크기와 제약을 추가하기 용이함을 보였다.
상세 분석
이 연구는 단백질 3차원 구조 예측을 전통적인 물리‑화학 기반 샘플링이 아닌 선언적 프로그래밍 관점에서 접근한다는 점에서 혁신적이다. 먼저, 저자들은 PDB와 같은 공개 구조 데이터베이스에서 고정 길이(보통 3~9잔기) 조각을 추출하고, RMSD와 토폴로지 유사도를 기준으로 클러스터링한다. 각 클러스터는 출현 빈도와 대표 구조를 메타데이터로 보유하며, 이는 조각 선택 시 확률적 가중치로 활용된다.
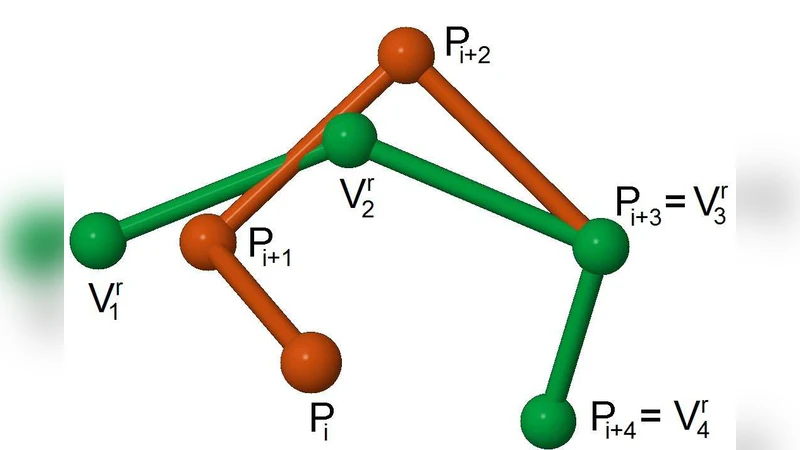
조각을 조합해 전체 단백질을 재구성하는 과정은 변수(조각 인덱스, 회전·이동 파라미터)와 제약(연결 부위의 거리·각도 일치, 충돌 방지, 사슬 연속성)으로 구성된 제약 만족 문제(CSP)로 변환된다. 여기서 CLP(FD) 엔진을 이용해 정수형 변수와 실수형 제약을 동시에 다루며, 백트래킹과 도메인 축소를 통해 탐색 공간을 급격히 감소시킨다.
에너지 모델은 기존의 통계적 잠재 에너지(예: DFIRE, DOPE)를 Ca‑측쇄 중심 모델에 맞게 재정의한다. 각 조각 사이의 비접촉 상호작용과 내부 구성을 합산해 전역 에너지 점수를 산출하고, 이는 목표 함수로 설정된다. 탐색 단계에서는 “large neighboring search”라 명명된 전략을 적용한다. 현재 해의 조각 하나를 무작위로 교체하거나, 인접 조각 집합을 동시에 재배치하는 큰 규모의 이웃을 생성해, 메타휴리스틱(예: 탭우 탐색)과 결합해 지역 최적에 빠지는 것을 방지한다.
실험에서는 50여 개의 중간 크기(100~200잔기) 단백질을 대상으로, 기존 fragment assembly 기반 도구와 비교했을 때 RMSD 감소와 계산 시간 단축을 동시에 달성했다. 특히, 조각 클러스터링 단계에서 빈도 기반 가중치를 도입함으로써 흔히 발생하는 “over‑sampling” 문제를 완화하였다.
한계점으로는 조각 길이와 클러스터 수에 따라 메모리 사용량이 급증할 수 있으며, 현재 구현은 단일 코어 환경에 최적화돼 병렬 확장성이 제한적이다. 또한, Ca‑측쇄 중심 모델은 사이드 체인의 세밀한 상호작용을 충분히 반영하지 못해, 활성 부위 예측 등 정밀도가 요구되는 응용에는 추가적인 후처리가 필요하다.
향후 연구 방향은 가변 길이 조각 도입, 사이드 체인 원자 모델링, 그리고 GPU 기반 CLP 엔진을 활용한 대규모 병렬 탐색이다. 이러한 확장은 선언적 모델의 장점을 유지하면서도 예측 정확도와 효율성을 동시에 향상시킬 것으로 기대된다.
{# ── Original Paper Viewer ── #}
댓글 및 학술 토론
Loading comments...
의견 남기기