통계적 평균 차이를 이용한 계통수 불일치 검정
초록
본 논문은 유전자 서열 정렬로부터 얻은 두 개의 계통수 분포가 통계적으로 유의하게 다른지를 검정하는 새로운 방법을 제시한다. 계통수를 고차원 특징 공간(분할·쿼터트)에 임베딩하고, 평균 벡터 간 차이를 커널 기법으로 효율적으로 계산한다. 부트스트랩을 통해 p값을 추정하며, 시뮬레이션과 실제 데이터(다람쥐·이, 초본·내생균, 균류 유전자)에서 방법의 유효성을 확인한다.
상세 분석
이 연구는 전통적인 ‘점 추정’ 방식이 아닌, 입력 정렬이 생성하는 계통수들의 전체 확률분포를 비교한다는 점에서 혁신적이다. 먼저 각 정렬에 대해 부트스트랩 혹은 베이즈 샘플링을 통해 다수의 계통수를 생성하고, 이를 분할(split) 혹은 쿼터트(quartet)와 같은 고차원 특징 벡터로 변환한다. 이러한 임베딩은 계통수 간 거리 계산을 선형 대수적 연산으로 단순화시키지만, 차원 수가 급격히 증가할 위험이 있다. 이를 해결하기 위해 저자들은 ‘커널 트릭’을 도입한다. 실제 특징 공간에 직접 매핑하지 않고, 두 평균 벡터 사이의 내적을 커널 함수(예: 선형·다항식 커널)로 대체함으로써 계산 복잡도를 크게 낮춘다.
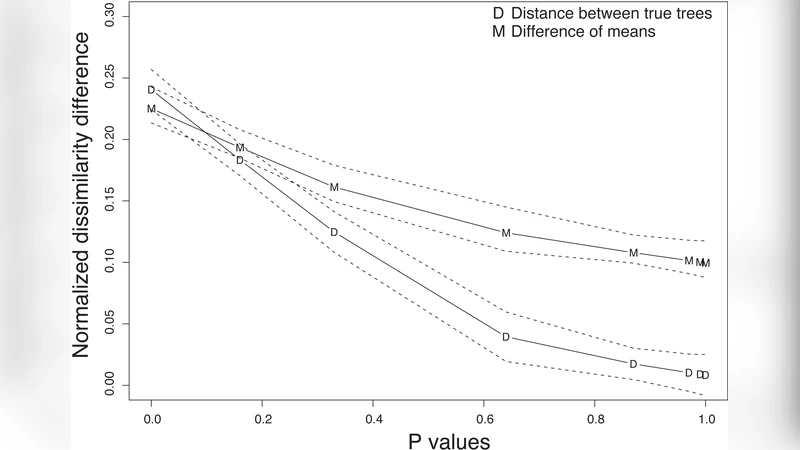
평균 차이 검정은 두 평균 벡터의 유클리드 거리 혹은 Mahalanobis 거리와 같은 통계량을 정의하고, 부트스트랩된 정렬 컬럼을 재샘플링하여 귀무분포를 구축한다. 이 과정에서 p값은 관측된 거리보다 큰 거리 비율로 추정된다. 시뮬레이션에서는 종 간 불일치 정도를 조절한 공동 진화(coalescent) 모델을 사용해, 종 트리와 유전자 트리 사이의 차이가 클수록 검정력이 상승함을 확인했다.
실제 데이터 적용에서는 (1) 다람쥐와 이의 공생 관계, (2) 초본 식물과 내생균, (3) 동일 균류 게놈 내 서로 다른 유전자군을 대상으로, 기존 방법으로는 포착하기 어려운 미세한 불일치를 성공적으로 탐지했다. 특히 수평 유전자 전달이나 유전체 재배열과 같은 비정상적 사건을 통계적으로 뒷받침할 수 있었다.
제안된 툴킷 ‘Phylotree’는 파이썬 기반으로, 정렬 입력 → 부트스트랩 → 임베딩 → 커널 거리 계산 → p값 산출까지 전 과정을 자동화한다. 그러나 고차원 특징 공간의 선택이 결과에 민감하며, 커널 파라미터 튜닝이 필요하다는 제한점이 있다. 또한 부트스트랩 반복 수가 충분히 크지 않으면 p값의 변동성이 커질 수 있다. 향후 연구에서는 차원 축소(예: 랜덤 프로젝션)와 베이즈 모델을 결합해 검정의 강건성을 높이는 방안을 모색할 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기