HP 사이드체인 모델 최적 구조의 동치 클래스
초록
본 논문은 수소성-극성(HP) 격자 단백질 모델에 측쇄를 포함시켜, 에너지 최소화에 의해 발생하는 방대한 최적 구조들을 동치 관계로 묶는다. 제안된 동치 클래스는 수소성 잔기의 배치만을 고려해 구조를 구분하며, 효율적인 대표 구조 열거 알고리즘을 제시한다. 이를 통해 구조 탐색 비용을 크게 낮추고, 단백질 설계·진화 연구에 활용할 수 있다.
상세 분석
HP 모델은 단백질을 격자 상의 사슬로 단순화하고, 잔기를 친수성(H)과 소수성(P) 두 종류로 구분한다. 기존 연구에서는 소수성 잔기 간의 접촉 수를 최소 에너지 함수로 사용했으나, 이 함수는 H‑H 접촉만을 고려하기 때문에 동일한 H‑H 접촉 패턴을 갖는 수천, 수만 개의 구조가 동일한 에너지를 갖는다. 이러한 구조 과잉은 시뮬레이션·열거 비용을 급격히 증가시키며, 실제 생물학적 의미를 추출하기 어렵게 만든다. 논문은 이 문제를 ‘동치 관계’를 도입함으로써 해결한다. 구체적으로, 두 구조가 동일한 H‑H 접촉 집합을 가질 경우 같은 동치 클래스에 속한다는 정의를 내렸다. 이는 에너지 함수와 직접적인 상관관계를 가지며, 클래스 내부의 구조들은 에너지적으로 구분되지 않는다.
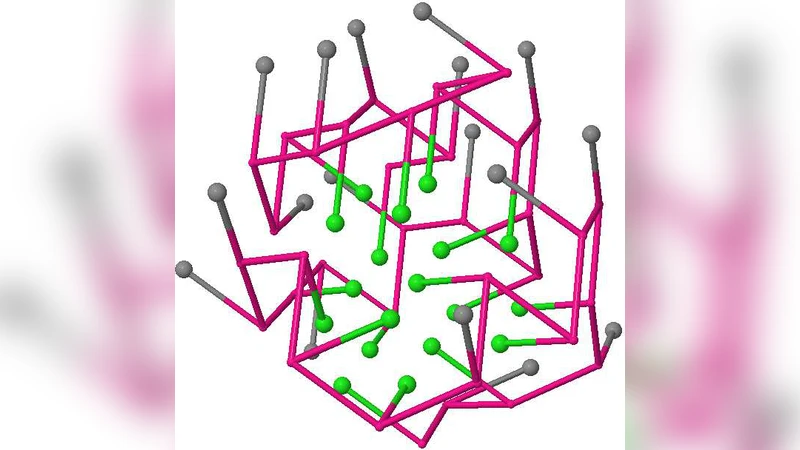
측쇄를 포함한 확장 모델에서는 각 아미노산이 백본과 측쇄 두 개의 격자 점으로 표현된다. 이때 백본은 사슬 연결을 보장하고, 측쇄는 H/P 특성을 부여한다. 논문은 측쇄 위치가 변해도 백본이 동일하면 같은 동치 클래스로 묶을 수 있음을 보인다. 이를 위해 ‘핵심 구조(core structure)’라는 개념을 도입했으며, 핵심 구조는 백본의 격자 좌표 집합으로 정의된다. 핵심 구조가 동일하면, 측쇄 배치가 달라도 에너지값은 변하지 않는다.
알고리즘적으로는 먼저 백본을 완전 탐색하거나 동적 계획법을 이용해 모든 가능한 백본을 열거한다. 각 백본에 대해 H‑H 접촉을 계산하고, 이를 해시값으로 변환해 동치 클래스 식별자를 만든다. 이후 각 클래스에 대해 대표 구조를 하나씩 선택한다. 대표 구조 선택 시, 측쇄 배치를 최소화하거나, 특정 생물학적 제약(예: 접촉 거리, 회전 대칭) 등을 추가 적용할 수 있다. 이 과정은 기존의 전체 구조 열거에 비해 메모리와 시간 복잡도가 크게 감소한다. 실험 결과, 30‑길이 이상의 서열에 대해 기존 방법이 수십억 개의 구조를 생성하는 반면, 제안된 방법은 수천 개의 동치 클래스만을 도출하였다.
또한 논문은 동치 클래스가 단백질 설계와 진화 시뮬레이션에 유용함을 입증한다. 설계 단계에서는 목표 에너지 이하의 구조를 찾을 때, 클래스 대표만을 검사하면 충분하므로 탐색 공간이 급격히 축소된다. 진화 시뮬레이션에서는 변이 후 구조가 기존 클래스에 속하는지 여부를 빠르게 판단함으로써, 중복된 계산을 방지한다. 이러한 장점은 특히 대규모 서열 집합이나 복잡한 측쇄 상호작용을 고려해야 하는 연구에 큰 도움이 된다.
결론적으로, 논문은 HP 모델에 측쇄를 포함한 경우에도 에너지 기반 동치 관계를 정의하고, 효율적인 열거·대표 선택 알고리즘을 제공함으로써 구조 탐색의 실용성을 크게 향상시켰다. 이는 향후 격자 모델을 이용한 단백질 설계, 구조 예측, 진화 분석 등에 폭넓게 적용될 수 있는 기반을 마련한다.
댓글 및 학술 토론
Loading comments...
의견 남기기