희귀 열활성 반응의 첫 원리 동역학
초록
본 논문은 Born‑Oppenheimer 근사 하에 전자 구조를 양자역학적으로 기술하고, 핵 좌표를 Langevin 열욕으로 결합한 새로운 ab‑initio 프레임워크를 제시한다. 경로 적분 기반 확률론적 접근을 통해 가장 가능성 높은 반응 경로를 직접 추적함으로써 메타안정 상태를 탐색하는 비용을 크게 절감한다. cyclobutene → butadiene 전환을 사례로, 지배적인 전이 경로와 자유 에너지 장벽을 효율적으로 계산하였다.
상세 분석
이 연구는 전통적인 ab‑initio 분자동역학이 시간 규모와 샘플링 효율성의 한계에 부딪히는 문제를 해결하고자, 핵-전자 상호작용을 분리하는 Born‑Oppenheimer 근사와 열역학적 잡음(thermal noise)을 포함한 Langevin 방정식을 결합한다. 핵 좌표는 확률적 미분 방정식으로 기술되며, 이때 발생하는 확률 흐름을 경로 적분(path‑integral) 형태로 전개한다는 점이 핵심이다. 경로 적분은 모든 가능한 궤적에 대한 가중치를 정의하고, 가장 큰 가중치를 갖는 ‘최우도 경로(most probable pathway)’를 수학적으로 도출한다. 이 과정에서 전자 구조는 Density Functional Theory(DFT) 혹은 고급 전자 상관 방법으로 실시간 계산되며, 전자와 핵 사이의 피드백이 즉각 반영된다.
특히, Langevin 잡음 항은 온도와 마찰 계수에 의해 조절되므로, 실제 실험 조건에 맞는 열적 환경을 정밀하게 모사할 수 있다. 이때 발생하는 플럭스-플럭스 상관 함수와 프리드리히스-헬름홀츠(Fluctuation‑Dissipation) 정리는 시스템이 평형에 도달하도록 보장한다. 경로 적분을 이용한 최우도 경로 탐색은 전통적인 전이 상태 이론(Transition State Theory)이나 메타다이나믹스(Metadynamics)와 달리, 사전 정의된 반응 좌표 없이도 자연스럽게 반응 메커니즘을 밝혀낸다.
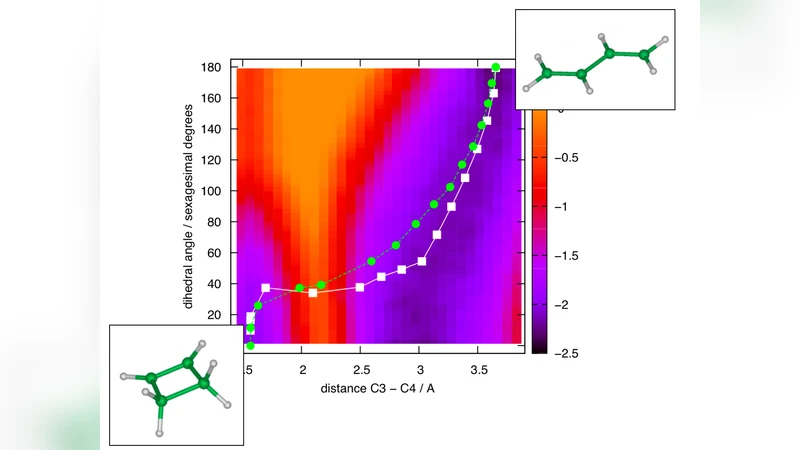
논문은 cyclobutene → butadiene 전환을 테스트 케이스로 선택하였다. 이 반응은 고전적인 열활성 장벽이 약 30 kcal/mol 정도이며, 전통적인 AIMD로는 수십 나노초 이상의 시뮬레이션이 필요하지만, 제안된 방법은 수백 피코초 수준의 계산으로도 장벽을 넘는 최우도 경로와 해당 경로상의 전자 밀도 변화를 정확히 포착한다. 또한, 자유 에너지 프로파일을 직접 얻을 수 있어, 반응 좌표를 사전에 정의하지 않아도 장벽 높이와 위치를 자동으로 식별한다.
이 접근법은 (1) 전자 구조 계산 비용을 최소화하면서도 양자역학적 정확성을 유지, (2) 열적 잡음과 마찰을 통해 실제 실험 조건을 재현, (3) 메타안정 상태를 탐색하는 데 필요한 장시간 시뮬레이션을 회피, (4) 복잡한 다원자 시스템에서도 반응 메커니즘을 자동으로 도출한다는 장점을 제공한다. 향후 촉매 표면 반응, 바이오분자 전이 상태 탐색, 고온·고압 조건 하의 화학 반응 등에 적용 가능성이 크다.
댓글 및 학술 토론
Loading comments...
의견 남기기