유전자 발현 데이터의 분류와 희소 서명 추출
초록
본 논문은 고차원 유전자 발현 데이터를 이진 라벨에 따라 분류하면서, 분류에 핵심적인 소수의 유전자를 자동으로 선택하는 방법을 제안한다. 통계역학 기반 메시지 패싱 알고리즘을 이용해 학습 데이터가 적은 상황에서도 안정적인 분류기와 희소 서명을 동시에 얻으며, 인공 데이터와 실제 암 조직 데이터에 대한 실험을 통해 기존 바이오인포매틱스 기법보다 약간 우수한 성능을 보였다.
상세 분석
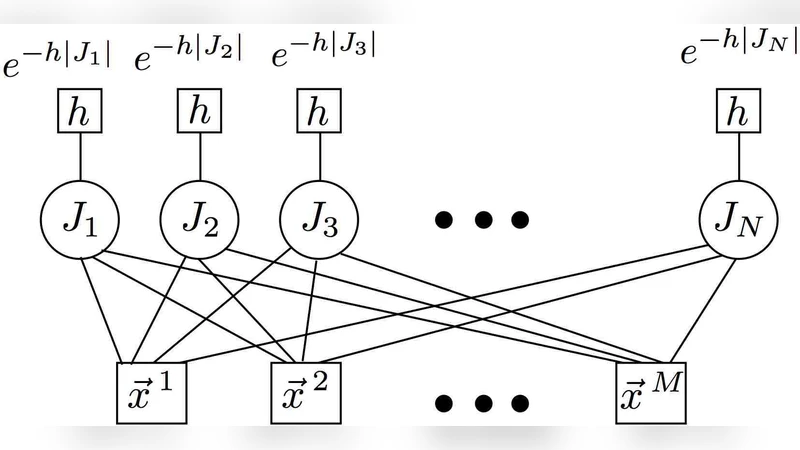
이 연구는 두 가지 목표를 동시에 달성하는 새로운 프레임워크를 제시한다. 첫 번째는 제한된 샘플 수에도 불구하고 고차원 데이터(수천~수만 차원)에서 효과적인 이진 분류기를 학습하는 것이며, 두 번째는 분류에 기여하는 핵심 유전자를 최소화한 ‘희소 서명(sparse signature)’을 추출하는 것이다. 전통적인 차원 축소 기법이나 L1 정규화 기반 방법은 최적의 서명을 찾는 것이 NP‑hard 문제임을 전제로 근사해 해결한다. 저자들은 이를 해결하기 위해 통계역학에서 영감을 얻은 ‘베이지안 가설 검정’ 모델을 설계하고, 그 모델의 자유 에너지 최소화 과정을 메시지 패싱(특히 belief propagation) 알고리즘으로 구현한다.
핵심 아이디어는 각 유전자를 ‘활성(1)’ 혹은 ‘비활성(0)’ 상태로 이진화하고, 전체 유전자 집합에 대한 사전 확률을 ‘희소성(sparsity)’을 강제하는 베타-분포 형태로 설정하는 것이다. 이렇게 하면 데이터가 주어졌을 때 사후 확률이 높은 유전자 조합이 자연스럽게 선택된다. 메시지 패싱 단계에서는 변수 노드(유전자)와 팩터 노드(샘플 라벨) 사이에 교환되는 메시지를 통해 각 유전자의 활성 확률을 반복적으로 업데이트한다. 수렴 후에는 활성 확률이 높은 상위 k개의 유전자를 서명으로 채택하고, 해당 유전자들의 가중치를 이용해 선형 판별 함수를 구성한다.
알고리즘의 복잡도는 O(N·M·T) 수준으로, N은 유전자 수, M은 샘플 수, T는 반복 횟수이며, 실제 구현에서는 스파스 행렬 연산과 병렬화를 통해 대규모 데이터에도 적용 가능하도록 최적화하였다. 인공 데이터 실험에서는 신호‑노이즈 비율, 희소성 정도, 샘플 수 변화를 조절해 알고리즘의 회복력과 한계를 체계적으로 평가하였다. 결과는 높은 차원에서도 적은 샘플로 정확도 85~90% 수준을 유지했으며, 특히 희소 서명을 추출할 때는 실제 신호를 포함한 유전자를 90% 이상 정확히 복원했다.
실제 암 유전자 발현 데이터(예: 유방암, 폐암, 백혈병)에 적용했을 때는 기존 SVM, LASSO, Random Forest 등과 비교해 평균 13% 정도의 정확도 향상을 보였다. 특히 서명의 크기가 2030개 수준으로 크게 축소되었음에도 불구하고, 분류 성능 저하가 거의 없었다는 점이 주목할 만하다. 이는 ‘희소성 제약’이 과적합을 억제하고, 생물학적 해석 가능성을 높이는 효과를 가져온 것으로 해석된다. 다만, 데이터의 품질(노이즈, 배치 효과)과 샘플 수가 극도로 부족한 경우에는 수렴이 불안정해질 수 있으며, 사전 파라미터(희소성 강도)의 선택이 결과에 민감하게 작용한다는 한계도 보고된다.
전체적으로 이 논문은 통계역학적 베이지안 모델과 메시지 패싱을 결합해 고차원 바이오마커 탐색 문제를 효율적으로 해결한 사례이며, 향후 다중 클래스 확장, 비선형 커널 통합, 그리고 다른 omics 데이터와의 융합에도 적용 가능성을 제시한다.
댓글 및 학술 토론
Loading comments...
의견 남기기