CLeFAPS: 형태문자 기반 고속 유연 단백질 구조 정렬
초록
CLeFAPS는 기존 강체 정렬 프레임워크인 CLePAPS를 기반으로, 파라미터 자동 조정, 다중 대응 집합 구축, 벡터 기반 연장 절차 등 세 가지 유연성을 도입한 빠른 단백질 구조 정렬 알고리즘이다. HOMSTRAD와 SABmark 벤치마크에서 경쟁 알고리즘과 비교했을 때 정밀도·재현율 모두 동등하거나 우수하면서, 실행 시간은 기존 방법의 1/50~1/150 수준으로 크게 단축된다.
상세 분석
CLeFAPS는 “Conformational Letters”(CL)라는 이산적인 구조 표현을 활용한다는 점에서 기존 좌표 기반 정렬과 차별화된다. CL은 ϕ/ψ 각을 사전 정의된 17개의 문자로 변환해, 서열과 유사하게 문자열 매칭을 수행할 수 있게 한다. 이 접근법은 거리 계산에 비해 연산량이 적고, 구조적 잡음에 강인한 특성을 제공한다.
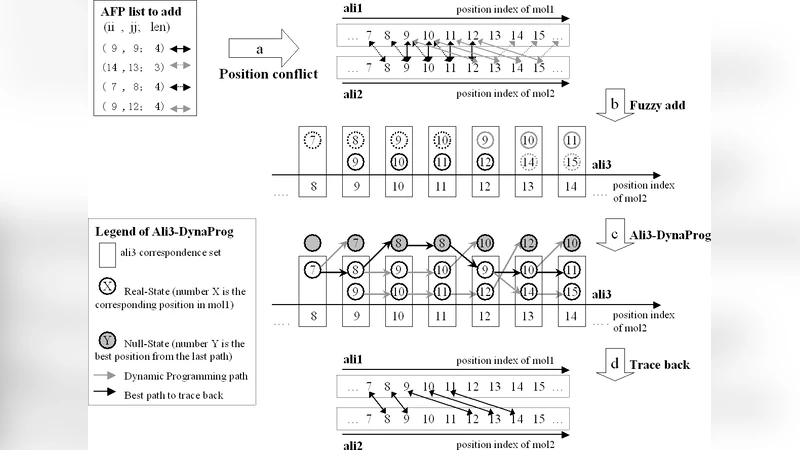
알고리즘의 핵심 유연성은 세 단계로 구분된다. 첫째, 파라미터 자동 적응은 입력 단백질의 길이에 비례해 거리 임계값(δ), 최소 AFP 길이(Lmin) 등을 동적으로 설정한다. 이는 짧은 단백질에서는 민감도를 높이고, 긴 단백질에서는 과도한 AFP 생성을 억제한다. 둘째, AFP를 “one‑to‑many” 대응 집합에 삽입함으로써, 기존 CLePAPS가 수행하던 위치 충돌 검사를 생략한다. 이는 동일 좌표에 여러 AFP가 겹칠 수 있게 하여, 구조적 변형을 자연스럽게 포착한다. 셋째, 연장 단계에서는 전통적인 거리 기반 점수 대신, 두 AFP 사이의 방향 벡터 차이를 이용한 스코어링을 적용한다. 벡터 차이는 회전·전이 변환에 대해 불변성을 가지므로, 실제 구조적 ‘굽힘’이나 ‘비틀림’ 없이도 연속적인 정렬을 확장할 수 있다.
성능 평가에서는 두 종류의 벤치마크를 사용하였다. HOMSTRAD은 고유사 단백질 쌍을, SABmark은 저유사·다양한 접힘을 포함한다. CLeFAPS는 TM-score와 RMSD 기준에서 대부분의 경우 기존 강체 정렬(TMalign, DALI)과 유연 정렬(MUSTANG, FATCAT)보다 동등하거나 약간 우수한 결과를 보였다. 특히 SABmark에서의 판별 테스트에서는 높은 특이도와 민감도를 유지하면서, 평균 실행 시간이 0.03~0.06초 수준으로, 경쟁 알고리즘의 1/100 이상 빠른 속도를 기록했다.
이러한 결과는 CL 기반 문자열 매칭이 구조 정렬의 근본적인 복잡도를 크게 낮추고, 파라미터 자동 적응과 다중 대응 집합이 실제 생물학적 변형을 효과적으로 모델링한다는 점을 시사한다. 또한, 벡터 기반 연장은 거리 기반 스코어링이 갖는 ‘임계 거리’ 문제를 회피해, 구조적 잡음에 대한 강인성을 높인다. 향후 다중 구조 정렬이나 대규모 구조 데이터베이스 검색에 적용하면, 고속·고정밀 정렬 엔진으로서 실용성이 크게 확대될 전망이다.
댓글 및 학술 토론
Loading comments...
의견 남기기