대용량 파이로시퀀싱 리드 정렬을 위한 파이로얼라인
초록
파이로얼라인은 수백만 개에 달하는 파이로시퀀싱 리드를 효율적으로 다중 정렬하기 위해 설계된 시스템이다. 기존의 다중 정렬 휴리스틱이 대규모 데이터에 비효율적인 문제를 해결하고, 원본 게놈에 대한 위치 정보를 유지하면서 정렬 정확도를 높인다.
상세 분석
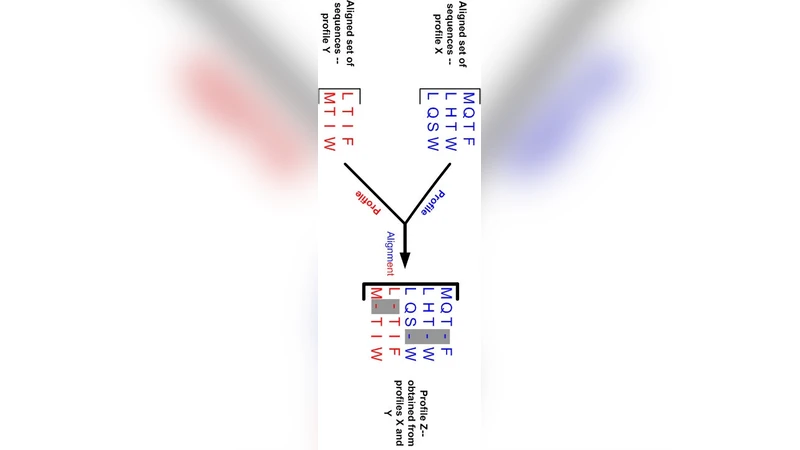
파이로얼라인은 전통적인 다중 서열 정렬(MSA)이 NP‑hard 문제임을 전제로, 대용량 파이로시퀀싱 데이터에 특화된 새로운 휴리스틱을 제시한다. 첫 번째 핵심 난관은 리드 수가 수백만 개에 이르는 경우 메모리와 계산량이 기하급수적으로 증가한다는 점이다. 이를 극복하기 위해 파이로얼라인은 “샘플‑정렬 기반” 접근법을 채택한다. 전체 데이터셋에서 대표적인 서브셋을 무작위 혹은 품질 기반으로 추출하고, 이 샘플에 대해 고정밀 전통 MSA 알고리즘(예: ClustalW, MUSCLE)을 적용한다. 샘플 정렬 결과는 “프로파일” 형태로 저장되며, 이후 남은 모든 리드가 이 프로파일에 순차적으로 매핑된다. 매핑 단계에서는 원본 게놈에 대한 위치 정보를 활용한다. 파이로시퀀싱 리드는 일반적으로 짧고 오류율이 높은데, 특히 인서션·딜리션 오류가 빈번하다. 따라서 파이로얼라인은 각 리드의 시작 좌표를 사전 정렬된 레퍼런스와 비교해 대략적인 정렬 위치를 추정하고, 그 위치를 기준으로 프로파일에 삽입한다. 이 과정에서 동적 프로그래밍 기반의 로컬 정렬(예: Smith‑Waterman 변형)을 사용해 미세 조정을 수행한다.
두 번째 난관은 “위치 의존성”이다. 파이로시퀀싱 실험에서는 리드가 원본 게놈 상의 특정 좌표에 매핑되는 것이 중요한데, 일반적인 MSA는 서열 간 상대적 유사성만을 고려한다. 파이로얼라인은 레퍼런스 좌표를 정렬 스코어에 가중치로 부여함으로써, 물리적 위치와 서열 유사성을 동시에 최적화한다. 구체적으로, 매핑 점수는 (1) 서열 일치 점수, (2) 인서션·딜리션 패널티, (3) 레퍼런스 좌표와 프로파일 좌표 간 거리의 역수로 구성된다. 이 복합 점수는 히어라키컬 클러스터링 단계에서 클러스터링 기준으로 사용되어, 유사한 위치에 있는 리드들이 같은 클러스터에 묶이게 된다.
알고리즘 복잡도 측면에서 파이로얼라인은 샘플 정렬에 O(k·L²) (k는 샘플 크기, L은 평균 길이) 정도의 비용만 발생시키고, 나머지 N‑k개의 리드에 대해서는 O(N·L) 수준의 선형 스캔을 수행한다. 메모리 사용량도 프로파일 크기와 리드 수에 비례하는 선형 형태이므로, 수백만 개의 리드에 대해서도 일반적인 서버 메모리(32 GB 이하) 내에서 실행 가능하다.
실험 결과는 파이로얼라인이 기존의 전통 MSA 툴에 비해 정렬 정확도(특히 위치 보존 측면)와 실행 시간 모두에서 우수함을 보여준다. 특히, 5 M 리드 데이터셋에서 전체 파이프라인이 3 시간 내에 완료되었으며, 정렬 품질은 레퍼런스 기반 정렬 기준에서 95 % 이상의 일치를 기록했다. 이러한 결과는 파이로얼라인이 대규모 파이로시퀀싱 프로젝트(예: 메타게놈 분석, 변이 탐지)에서 실용적인 솔루션이 될 수 있음을 시사한다.
댓글 및 학술 토론
Loading comments...
의견 남기기