다중 서열 정렬 시스템 종합 고찰
초록
본 논문은 현재까지 개발된 주요 다중 서열 정렬(MSA) 시스템들을 포괄적으로 정리하고, 각 시스템이 채택한 알고리즘, 구현 절차, 그리고 한계점을 비교한다. 또한 정렬 품질을 평가한 결과를 제시하며, 서열 종류·길이·진화적 거리 등에 따라 어느 시스템이 최적의 선택이 되는지를 논의한다.
상세 분석
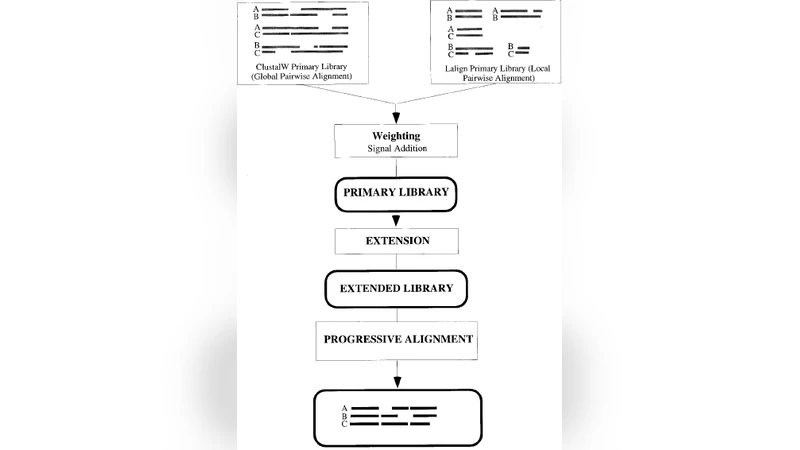
다중 서열 정렬은 생물학적 데이터 해석의 핵심 단계이며, 본 논문은 기존 MSA 도구들을 크게 세 가지 범주(진화 기반, 점수 기반, 혼합형)로 구분한다. 진화 기반 방법으로는 Clustal W, MUSCLE, MAFFT 등이 소개되며, 이들 모두 계통수 정보를 활용해 점진적으로 서열을 병합한다. 특히 MAFFT는 FFT 기반 가속을 통해 대규모 데이터셋에서도 높은 정확도를 유지한다는 점이 강조된다. 점수 기반 방법으로는 T-Coffee와 ProbCons가 대표적인데, 이들은 다중 정렬 스코어를 최적화하기 위해 동적 계획법이나 확률적 모델을 적용한다. T-Coffee는 여러 개별 정렬 결과를 통합해 신뢰도를 높이는 ‘합성 정렬’ 방식을 도입했으며, ProbCons는 은닉 마르코프 모델(HMM)을 이용해 서열 간 상관관계를 정밀하게 추정한다. 혼합형 접근법으로는 PRANK와 PASTA가 논의되는데, PRANK는 삽입·삭제 사건을 진화적 사건으로 모델링해 과도한 갭을 방지하고, PASTA는 반복적인 분할·정복 전략을 통해 메모리 사용량을 최소화한다. 논문은 각 알고리즘의 시간·공간 복잡도를 표로 정리하고, 특히 대용량 NGS 데이터에 대한 확장성을 평가한다. 제한점으로는 진화 기반 방법이 진화 모델 선택에 민감하고, 점수 기반 방법이 계산 비용이 크게 증가한다는 점을 지적한다. 또한, 정렬 품질 평가지표인 SP-score, TC-score, 그리고 구조 기반 RMSD를 활용한 실험 결과를 통해, 단백질 구조 보존 영역에서는 진화 기반 도구가 우수하지만, 비보존 영역이나 짧은 DNA 서열에서는 점수 기반 도구가 더 높은 정확도를 보인다는 중요한 인사이트를 제공한다. 마지막으로, 저자는 향후 연구 방향으로 딥러닝 기반 정렬 예측, 클라우드 환경에서의 병렬 처리, 그리고 사용자 정의 비용 함수 도입 등을 제안한다.
댓글 및 학술 토론
Loading comments...
의견 남기기