다중 검정에서 교란을 고려한 새로운 FDR 및 수축 추정 방법
본 논문은 유전학·통계학에서 제안된 교란 보정 기법들을 통합하고, 교란을 반영한 두 가지 새로운 다중 검정 절차를 제시한다. 첫 번째는 민감도 분석을 활용해 교란을 보정한 뒤 FDR(거짓 발견율) 추정 및 q‑값 계산을 수행하는 방법이며, 두 번째는 혼합 모델 기반의 수축 추정기를 이용해 π₀(진정한 영가설 비율)와 효과 크기를 동시에 추정한다. 프로스테이트 암 유전자 발현 데이터에 적용해 실효성을 검증한다.
저자: Debashis Ghosh
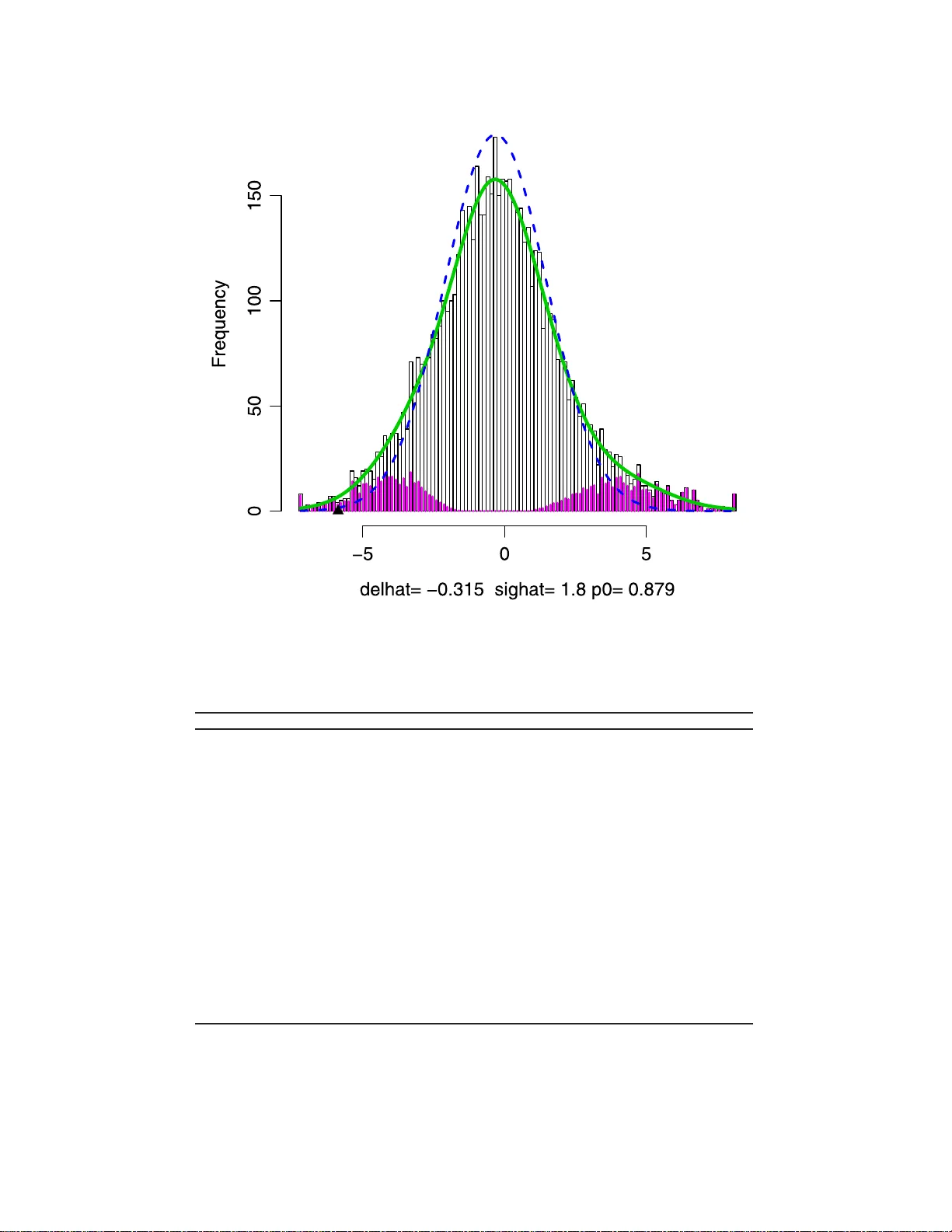
본 논문은 다중 가설 검정에서 교란(confounding) 문제가 발생할 경우 기존의 거짓 발견율(FDR) 제어 절차가 잘못된 영가설(null) 분포를 전제로 하여 오류를 일으킬 수 있음을 지적한다. 이를 해결하기 위해 저자는 먼저 전통적인 혼합 모델 f(t)=π₀f₀(t)+(1−π₀)f₁(t) 를 교란을 반영하도록 확장하여 f_C0(t)와 f_C1(t) 로 표기한다. 여기서 f_C0는 교란에 의해 평균·분산이 변형된 영가설 분포이며, f_C1은 대안 가설 분포가 동일하게 변형된다고 가정한다.
문헌에서 제시된 세 가지 교란 보정 방법—Devlin‑Roeder의 유전체 제어(genomic control), Pritchard‑Rosenberg의 잠재 군집 변수 모델, Efron의 경험적 영가설(empirical null)—을 동일한 혼합 모델 틀 안에 위치시켜 비교한다. Devlin‑Roeder는 보조 통계량 U₁,…,U_K 를 이용해 과분산(σ)만을 추정하고, 이를 통해 f₀를 스케일 조정한다. Pritchard‑Rosenberg은 잠재 변수 C를 추정해 각 군집 내에서 전통적인 영·대안 분포를 그대로 적용한다. Efron은 전체 통계량을 이용해 영가설 분포를 데이터 기반으로 추정하며, “zero‑matching” 가정을 통해 f₀와 f₁을 지수적 기울기로 연결한다.
이러한 기존 방법들의 장단점을 정리한 뒤, 논문은 두 가지 새로운 절차를 제안한다.
첫 번째는 Lin et al.의 민감도 분석(framework) 를 차용해, 관측되지 않은 교란 변수 C를 평균 μ_D와 회귀계수 γ 로 요약한다. 사용자가 γ와 μ₁−μ₀ 를 사전 지정하면 보정된 회귀계수 β̂₁를 직접 계산할 수 있다. 구체적으로, 원래 회귀식 E(X|D,C)=β₀+β₁D+γC 를 C가 정규분포(N(μ_D,1)) 라는 가정 하에 적분하면 β₁=β₁*−γ(μ₁−μ₀) 가 된다. 여기서 β₁*는 교란을 무시하고 추정한 계수이며, 이를 보정하면 교란을 반영한 Wald 통계량을 얻는다. 이렇게 얻은 p‑값을 기존 Storey의 q‑값 추정 절차 혹은 Benjamini‑Hochberg 절차에 그대로 투입함으로써 교란 보정과 FDR 제어를 일관되게 수행한다. 민감도 분석 단계에서 표준 오차가 변하지 않으므로, 검정 통계량은 기존과 동일하게 해석 가능하다.
두 번째 절차는 혼합 모델 기반의 수축 추정(shrinkage estimation) 을 도입한다. π₀와 효과 크기 θ를 동시에 추정하기 위해 James‑Stein형 수축 추정량을 사용하고, 이를 통해 “empirical Bayes” 형태의 사후 평균을 구한다. 구체적으로, EM 알고리즘을 이용해 f_C0와 f_C1을 추정하고, 각 검정 통계량에 대해 수축된 효과 추정값을 계산한다. 이 과정에서 π₀̂ 를 업데이트함으로써 전체 검정에서 영가설 비율을 보다 보수적으로 추정한다. 수축 추정은 특히 대규모 유전자 데이터에서 개별 검정 통계량이 고분산·편향을 보일 때, 전체 분포 정보를 활용해 각 검정의 신뢰도를 향상시킨다.
실증 분석에서는 전립선 암 마이크로어레이 데이터를 사용한다. 교란 변수로는 배치 효과와 조직 이질성을 가정하고, 민감도 분석 파라미터(γ, μ₁−μ₀)를 여러 시나리오에 걸쳐 변동시켰다. 결과적으로 교란 보정 전후의 p‑값 분포가 크게 달라짐을 확인했으며, 보정 후에는 더 많은 유전자가 FDR 0.05 이하로 검출되었다. 수축 추정 방법은 π₀̂ 를 보다 보수적으로 추정해 과도한 발견을 억제하면서도, 실제 의미 있는 유전자를 놓치지 않는 균형을 제공한다.
논문은 또한 교란을 명시적으로 모델링하고, 민감도 분석과 경험적 베이지안 수축 추정을 결합함으로써 기존 FDR 제어 방법의 한계를 극복한다는 점을 강조한다. 제안된 두 절차는 교란이 존재하는 고차원 생물학 데이터에 적용 가능하며, 통계적 검정의 정확도와 재현성을 동시에 향상시킬 수 있다. 마지막으로, 교란 보정이 필요한 상황에서 연구자는 보조 통계량의 존재 여부, 교란 파라미터의 사전 지정 가능성, 그리고 수축 추정의 계산 복잡성을 고려해 적절한 방법을 선택할 것을 권고한다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기