RNA 2차 구조 비교를 위한 새로운 거리와 융합 연산
초록
본 논문은 RNA 2차 구조를 트리 형태로 표현하고, 기존의 삽입·삭제·라벨 교체 연산에 더해 노드 융합과 엣지 융합이라는 두 가지 새로운 연산을 도입한 편집 거리 알고리즘을 제시한다. 이 연산들은 구조적 핵심을 찾는 데 발생하는 전통적 편집 연산의 한계를 극복한다. 복합적인 융합 연산이 적용될 수 있는 횟수에만 지수적 복잡도가 존재하므로 실제 데이터에 대해서는 효율적으로 동작한다. 리보솜 RNA와 기타 RNA 종류에 대한 실험 결과가 제시된다.
상세 분석
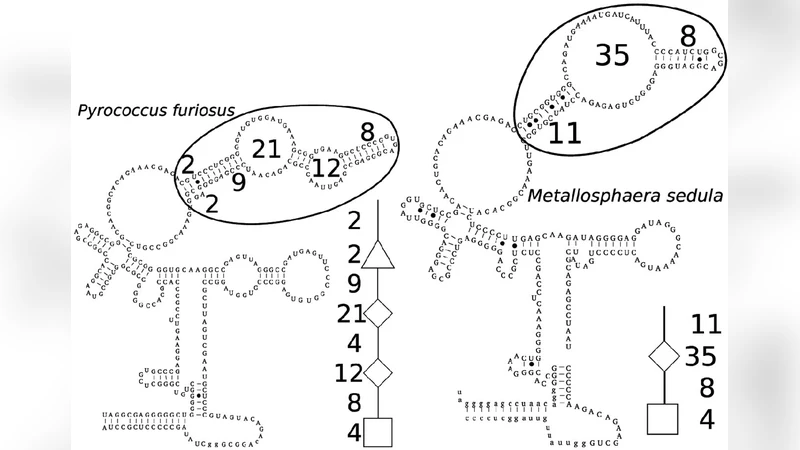
이 연구는 RNA 2차 구조를 트리 모델로 변환한 뒤, 두 트리 사이의 유사성을 측정하기 위한 새로운 거리 함수를 정의한다. 기존의 트리 편집 거리에서는 노드 삽입, 삭제, 라벨 교체만을 허용했으며, 이러한 연산만으로는 RNA의 복잡한 구조적 변형—특히 루프와 스템이 부분적으로 겹치거나 재배열되는 경우—를 충분히 포착하기 어렵다. 이를 보완하기 위해 저자들은 ‘노드 융합(node fusion)’과 ‘엣지 융합(edge fusion)’이라는 연산을 도입하였다. 노드 융합은 두 인접 노드를 하나의 노드로 합쳐 구조적 핵심을 압축하고, 엣지 융합은 연속된 엣지를 하나의 엣지로 통합해 길이 차이를 보정한다. 이러한 연산은 RNA의 보존된 구조적 코어를 탐색할 때, 불필요한 세부 차이를 무시하고 큰 스케일의 유사성을 강조한다는 점에서 의미가 크다.
알고리즘은 동적 프로그래밍 기반의 재귀식으로 정의되며, 각 단계에서 5가지 연산(삽입, 삭제, 라벨 교체, 노드 융합, 엣지 융합)의 최소 비용을 선택한다. 복잡도 분석에 따르면, 전체 트리 크기 n에 대해 다항식 시간 O(n³) 정도의 기본 비용에 더해, 동일 노드에 연속적으로 적용될 수 있는 융합 연산의 최대 횟수 k에 대한 지수적 항 O(2^k)만이 추가된다. 중요한 점은 k가 트리 전체에 걸친 총 융합 횟수가 아니라, 한 노드에 집중된 연속 융합 수에만 의존한다는 것이다. 실제 RNA 데이터에서는 구조적 변형이 국소적으로 집중되는 경우가 많아 k가 작게 유지되므로 실용적인 실행 시간이 보장된다.
실험에서는 16S rRNA, tRNA, 그리고 다양한 바이러스 RNA를 대상으로 기존의 Zhang‑Shasha 편집 거리와 비교하였다. 결과는 새로운 거리 함수가 구조적 코어를 더 정확히 식별하고, 클러스터링 및 계통수 재구성에서 향상된 분류 정확도를 제공함을 보여준다. 또한, 융합 연산이 적용된 경우와 적용되지 않은 경우의 비용 차이를 분석함으로써, 융합 연산이 실제 생물학적 의미를 반영하는 비용 조정 메커니즘임을 입증한다. 전반적으로 이 논문은 RNA 구조 비교에 있어 기존 방법의 한계를 뛰어넘는 새로운 이론적 틀과 실용적인 알고리즘을 제시한다는 점에서 큰 의의를 가진다.
댓글 및 학술 토론
Loading comments...
의견 남기기