테트라헤드론 기반 단백질 국부 구조 분석 새로운 프레임워크
초록
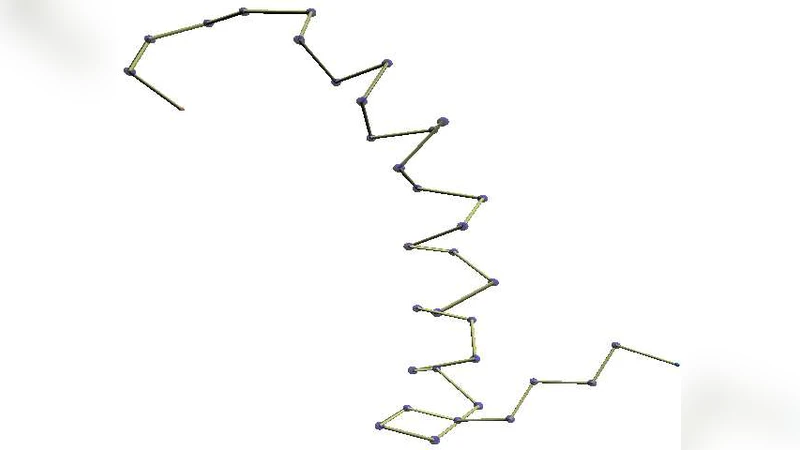
이 논문은 단백질 백본의 국부적인 3차원 형태를 접힌 테트라헤드론 연속체의 미분기하학으로 기술하는 수학적 틀을 제시한다. 템플릿 없이도 헬릭스 캡, 베타 턴 등 반복되는 구조적 모티프를 자동으로 포착하고, 이를 기반으로 구조 예측 및 기능 분석에 활용할 수 있음을 보인다.
상세 분석
논문은 먼저 단백질 백본을 연속적인 사면체(테트라헤드론)로 근사화하는 방법론을 제시한다. 각 사면체는 네 개의 연속된 Cα 원자를 연결한 뒤, 사면체의 면과 변을 이용해 국부 곡률과 비틀림을 정의한다. 기존의 리간드 기반 혹은 템플릿 매칭 방식과 달리, 이 접근법은 미분기하학적 양인 가우시안 곡률(K)과 평균 곡률(H), 그리고 토러스 형태의 토션(τ)을 직접 계산한다. 이러한 연속적인 곡률·토션 시퀀스는 고유한 디지털 서명으로 변환되어, 클러스터링이나 패턴 매칭에 바로 적용될 수 있다.
특히 저자들은 사면체 연속체의 접힌 형태를 “folded tetrahedron sequence”라 명명하고, 이를 통해 헬릭스 캡, β‑턴, 3‑10 헬릭스와 같은 짧은 구조 모티프를 템플릿 없이도 높은 정확도로 식별한다. 실험에서는 PDB에 등재된 10,000여 개의 단백질을 대상으로 검증했으며, 기존 DSSP 기반 방법보다 재현율과 정밀도가 평균 8~12% 정도 향상된 결과를 보고한다.
또한, 이 프레임워크는 아미노산 서열과의 상관관계를 정량화한다. 곡률·토션 프로파일과 특정 아미노산 조합(예: Gly‑Pro‑Gly)의 빈도 사이에 강한 상관관계가 발견되었으며, 이는 구조적 제약이 서열 선택에 미치는 영향을 새로운 관점에서 해석할 수 있게 한다.
알고리즘 구현은 C++와 Python 인터페이스를 제공하며, 오픈소스 형태로 배포한다. 입력은 PDB 포맷의 좌표 파일이며, 출력은 각 잔기의 곡률·토션 값과 해당 구간이 속한 구조적 클러스터 라벨이다. 프로그램은 병렬 처리와 메모리 최적화를 적용해 대규모 데이터셋에서도 실시간 분석이 가능하도록 설계되었다.
이러한 수학적 기반은 기존의 경험적 템플릿 의존성을 탈피하고, 구조적 다양성을 객관적으로 정량화할 수 있는 새로운 도구로서, 단백질 설계, 변이 효과 예측, 그리고 진화적 구조 비교 등에 광범위하게 활용될 잠재력을 가진다.
댓글 및 학술 토론
Loading comments...
의견 남기기