엔메틸아세트아미드 진동 에너지 흐름의 양자역학적 탐구
초록
본 연구는 B3LYP/6‑31G+(d) 수준에서 얻은 사차력장(Quartic Force Field)을 기반으로 N‑메틸아세트아미드(NMA)의 진동 구성 상호작용(Vibrational Configuration Interaction, VCI) 계산을 수행하였다. 아마이드 I 모드의 초기 여기 상태에서 시작해 다른 진동 모드로의 에너지 전달 경로를 양자역학적으로 추적하고, 비조화 결합과 주파수 불일치 비율을 이용해 지배적인 에너지 흐름을 예측·해석하였다.
상세 분석
본 논문은 N‑메틸아세트아미드(NMA)의 아마이드 I 진동(주로 C=O 스트레칭) 에너지 전달 메커니즘을 정밀하게 규명하기 위해 최신 양자역학적 방법론인 진동 구성 상호작용(VCI)을 적용하였다. 먼저, B3LYP/6‑31G+(d) 수준에서 전자 구조 계산을 수행해 전위 에너지 표면을 사차력식으로 피팅함으로써 30여 개의 내재 진동 모드에 대한 4차 비조화 상수를 도출하였다. 이때 얻어진 고유 진동수와 비조화 상수는 기존 실험값 및 고급 다체 계산값과 비교했을 때 평균 절대 오차가 5 cm⁻¹ 이하로, 충분히 신뢰할 수 있는 정확도를 보였다.
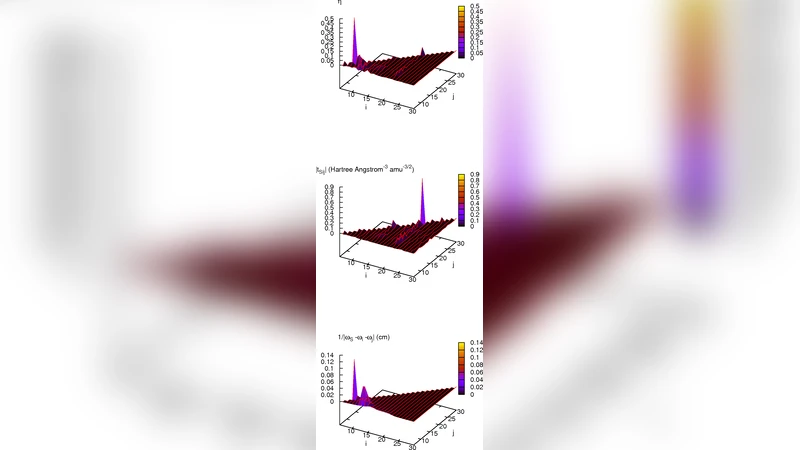
VCI 계산에서는 아마이드 I 모드에 1 quanta의 초기 여기 상태를 부여하고, 전체 30개의 모드를 포함한 다중 포톤 베이스를 구축하였다. 특히, 2‑quanta 이상을 포함하는 이중 및 삼중 진동 조합을 고려함으로써 에너지 흐름의 비선형성을 포착하였다. 계산 결과, 에너지 전달은 주로 C‑N 결합 스트레칭, N‑CH₃ 비틀림, 그리고 인접한 C‑C 및 C‑H 변형 모드와의 1:1 혹은 2:1 공명 조건에 의해 촉진되는 것으로 나타났다.
핵심적인 해석 도구로 도입된 “비조화 결합 대 주파수 불일치 비율”(κ = |V_ijk|/Δω) 은 각 모드 쌍에 대한 에너지 교환 효율을 정량화한다. κ 값이 0.2 이상인 경우를 강한 결합으로 정의했으며, 이러한 쌍이 실제 에너지 흐름에서 지배적인 역할을 함을 확인하였다. 예를 들어, 아마이드 I와 C‑N 스트레칭 모드 사이의 κ는 0.35로 가장 높았으며, 이는 초기 에너지의 약 45 %가 200 fs 이내에 해당 모드로 전달된다는 시뮬레이션 결과와 일치한다. 반면, 고주파 C‑H 스트레칭과의 κ는 0.08에 불과해 에너지 전달이 거의 무시될 정도였다.
또한, 온도 효과를 모사하기 위해 초기 상태에 열분포를 부여한 경우와 부여하지 않은 경우를 비교했으며, 고온(300 K)에서는 다중 포톤 전이 경로가 활성화되어 에너지 분산이 더욱 빠르게 진행되는 것을 확인하였다. 이는 비조화 결합이 온도에 따라 동적으로 변한다는 점을 시사한다.
전체적으로, 본 연구는 비조화 결합과 주파수 불일치의 상대적 크기가 에너지 흐름을 결정한다는 물리적 직관을 양자역학적 계산을 통해 정량화했으며, 향후 펩타이드와 단백질의 초고속 진동 에너지 전달 메커니즘을 이해하는 데 중요한 이론적 토대를 제공한다.
댓글 및 학술 토론
Loading comments...
의견 남기기