원자 확산 모델을 이용한 1D NMR 스펙트럼 기반 소분자 구조 규명
📝 원문 정보
- Title: Atomic Diffusion Models for Small Molecule Structure Elucidation from NMR Spectra
- ArXiv ID: 2512.03127
- 발행일: 2025-12-02
- 저자: Ziyu Xiong, Yichi Zhang, Foyez Alauddin, Chu Xin Cheng, Joon Soo An, Mohammad R. Seyedsayamdost, Ellen D. Zhong
📝 초록 (Abstract)
핵자기공명(NMR) 분광법은 소분자 구조를 규명하는 핵심 기술이며, 특히 새로운 천연물 및 임상 치료제 발굴에 필수적이다. 그러나 NMR 스펙트럼 해석은 시간과 전문 지식이 많이 요구되는 수작업 과정이다. 본 연구에서는 1차원 NMR 스펙트럼과 화학식을 입력으로 하여 미지 분자의 구조를 직접 예측하는 엔드투엔드 프레임워크 CHEFNMR을 제안한다. 구조 규명을 조건부 생성 문제로 정의하고, 비대칭 트랜스포머 기반 원자 확산 모델을 활용한다. 천연물에 흔히 나타나는 복잡한 작용기를 학습시키기 위해 111,000여 종 이상의 천연물에 대한 시뮬레이션 1D NMR 스펙트럼 데이터셋을 구축하였다. CHEFNMR은 어려운 천연물 화합물에 대해 65 % 이상의 정확도로 구조를 예측하며, 소분자 구조 규명의 자동화에 큰 진전을 제시한다. 코드와 데이터는 공개 URL에서 확인할 수 있다.💡 논문 핵심 해설 (Deep Analysis)
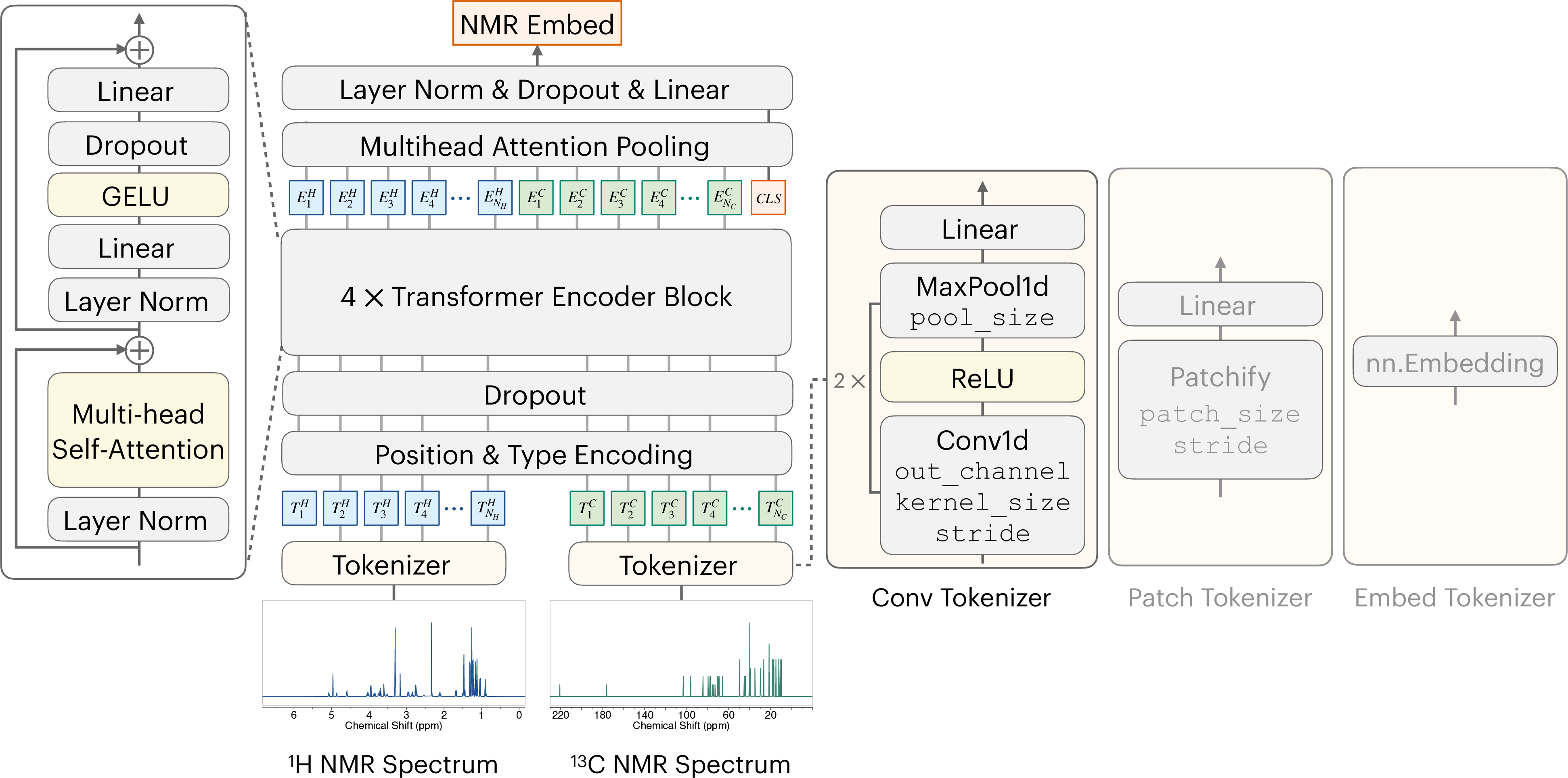
데이터 측면에서 저자들은 111 k개의 천연물에 대해 시뮬레이션 1D NMR 스펙트럼을 생성하였다. 이는 실제 실험 데이터의 부족을 보완하고, 다양한 작용기와 입체화학적 변이를 포괄하도록 설계된 점이 장점이다. 그러나 시뮬레이션 스펙트럼이 실제 실험 환경에서 발생하는 잡음, 용매 효과, 온도 변동 등을 완전히 반영하지 못한다는 한계도 존재한다. 향후 실제 실험 데이터와의 도메인 적응(domain adaptation) 연구가 필요하다.
성능 평가에서 65 % 이상의 정확도는 기존 방법 대비 현저히 높은 수치이며, 특히 ‘challenging natural product compounds’라 명시된 복잡한 구조군에서 좋은 결과를 보였다. 하지만 정확도라는 지표만으로는 구조 후보군의 다양성, Top‑k 정확도, 혹은 구조‑스펙트럼 일치도(chemical shift error) 등을 충분히 파악하기 어렵다. 논문에 제시된 정량적 분석이 제한적이므로, 향후 베이스라인과의 상세 비교, 오류 사례 분석, 그리고 실험실 검증을 통한 실용성 검증이 필요하다.
기술적 기여 외에도 CHEFNMR은 오픈소스 코드와 데이터셋을 공개함으로써 재현 가능성을 높였으며, 이는 학계와 산업계가 이 프레임워크를 확장하거나 다른 스펙트럼 유형(예: 2D NMR, HSQC)으로 확장하는 데 큰 도움이 될 것이다. 전반적으로 이 연구는 원자 확산 모델을 화학 구조 예측에 적용한 최초 사례 중 하나로, 향후 ‘자동화된 구조 규명’이라는 궁극적 목표에 한 걸음 더 다가가는 중요한 이정표라 할 수 있다.
📄 논문 본문 발췌 (Translation)
📸 추가 이미지 갤러리

