Autotuning T-PaiNN: Enabling Data-Efficient GNN Interatomic Potential Development via Classical-to-Quantum Transfer Learning
Machine-learned interatomic potentials (MLIPs), particularly graph neural network (GNN)-based models, offer a promising route to achieving near-density functional theory (DFT) accuracy at significantly reduced computational cost. However, their pract…
Authors: Vivienne Pelletier, Vedant Bhat, Daniel J. Rivera
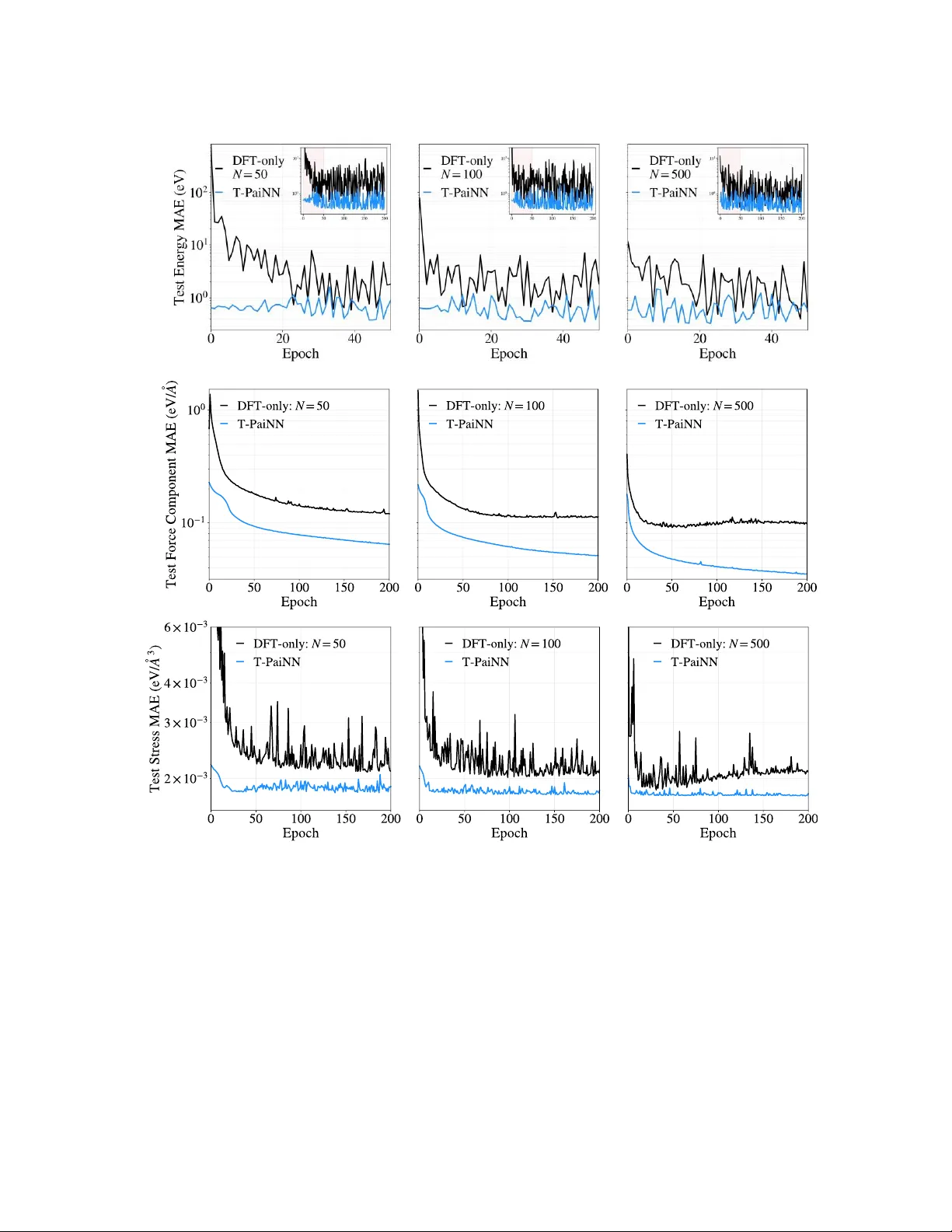
Pelletier et al, 20 26 ArXive Preprin t 1 Autotuning T-PaiNN: Enabling Data-Efficient GNN Interatomic Potential Development via Classical- to -Quantum Transfer Learning Vivienne Pelletier 1 , Vedant Bhat 2 , Daniel J. Rivera 2 , Steven A. Wilson 2 , Christopher L. Muhich 1,2 * 1 Materials Science & Engineering, School for the Engineering of Matter, Transport, & Energy, Arizona State University, 551 E. Tyler Mall, Tempe Arizona , 85287, USA 2 Chemical Engineering, School for the Engineering of Matter, Transport, & Energy, Arizona State University, 551 E. Tyler Mall, Tempe Arizona, 85287, USA * Corresponding Author Abstract Machine-learned interatomic potentials (MLIPs), particular ly graph neural network (GNN)-based models, oer a promising route to achieving near–density functional theory (DFT) accuracy at signicantly reduced comput ational cost. However , their p ractical d eployment is often limi ted by the lar ge volumes of expensive quantum mechanical training data required. In this work, we introduce a transfer learning fra mework, Transfer -PaiNN (T -PaiNN), that substantially improves the data eciency of GNN-MLIPs by lev eraging inexpensive classical force eld data. e approach consists of pretraining a PaiNN MLIP architecture on large-scale datasets generated from classical molecular simul ations, followed by ne-tuning (dubbed autotuning) using a comparatively small DFT dataset. W e demonstrate the eectiveness of autotuning T -PaiNN on both gas-phase molecular systems (QM9 da taset) and condensed-phase liquid water . Across all cases, T -PaiNN signicantl y outperforms models trained solely on DFT data, achieving order-of- magnitude reductions in mean absolute error while acce lerating training converge nce. For example, using the QM9 data set, error reductions of up to 25× are observe d in low-data regimes, while liquid wa ter simulations show improved predictions of ener gies, forces, and experimentally relevant p roperties such as density and di usion. ese gains arise from th e model’ s ability to learn general features of the potential energy surface from extensive classical sampling, which are subsequently rened to quantum accuracy . Overall, this work establishes transfe r l earning from classical force elds a s a pr actical and computationally ecient strategy for d eveloping high - accuracy , data-ecient GNN interatomic potent ials, enabling broader application of MLIPs to complex chemical systems. Pelletier et al, 20 26 ArXive Preprin t 2 1. Intr oduction e de velopment and use of machine l earned interatomic potentials (MLIPs) is rapidly e xpanding due to their promise for overcoming the chemical accuracy/computational speed tradeo that has dened computational atomistic modeling since its inception. [1] MLIPs are appealing surrogat e models for acquiring near-quantum mechanical, e.g. density functional theory (DFT), accuracy at near - classical molecular mechanics costs.[2] ML IPs have been developed as both stand-alone models, for example in molec ular dynamics simulations,[3] and as complimentary DFT accelerators, for example in geometry optimization and tra nsition state se arches.[4][5] Among the broad array of candidate machine learning architectures used for ML IPs, two predominant categories have emer ged: 1) Kernel-regression base d (Kernel-MLIP ), and 2) graph neural netwo rk (GNN),[6] due to their ability to broa dly reproduce DFT level accuracy , transferability , and size agnosticism. Kernel-MLIPs such as Gaussian Approximation Potentials [2] and their derivatives, e.g. V ASP ’s MLF Fs,[7] are appealing DFT surrogates because of their strong performance in the low training data regime. Kernel-MLIPs predict system ener gies by calculating the similarity between the atomic structure of int erest and all structure s in the training set via a similarity func tion , the kernel. Energy and forc e predictions of new structures are c onstructed from a learned linear combination of similarit ies to the training data. Forces and stresses a re calculated from the positional a nd c ell lattice vector analytica l derivatives of this procedure. e most p opular Ke rnel- MLIP methods employ Bayesian inference to provide an inbuilt self-assessment of their own uncertainty , thus enabling both retrospective assessment of calculation res ults and active learning “on-the-y” launching of DFT calculations. Kernel-MLIP memory demands scale poorly , typically as 2 , with tra ining set size .[8] Although this scaling may remain mana geable for narrowly tar geted inv estigation s, it becomes prohibitive when the objective is to tre at many dissimilar candidate molecules, solid materials, surface terminations, etc . While methods for r educing the memory d emands of Kernel-MLIPs exist, such as sparse inducing points and Nyström approximations, these approac hes only improve the unfavorable scaling from 2 to 2 , where is related to the sparsity of the approximation.[9] In practice, to maintain the necessary accuracy for MLI P applications, must be chosen so lar ge that the improvement in memory scaling is ultim ately modest.[8] us, alternative MLIP architectures must be used if broad applicability is desired. GNN -MLIPs predict mo lecular syst em energies and properties using a s eries of neural networks, and/or their derivatives (i.e. convoluti ons, perceptron, et c.).[10] Each atom is rst assigned a descriptor vector , typically initialized using its element type, then a GNN performs iterative updates of these embeddings based on interactions with neighboring atoms in the atom’ s chemical environment. Lastly , the atomic descriptors feed a NN that predicts each atom’ s individual contribution to the system ener gy (o r ot her desir ed prop erty). GNNs encompass a ra nge of architectures, such as SchNet,[10] Polarizable Atom Interaction Neural Network (PaiNN), [1 1] Pelletier et al, 20 26 ArXive Preprin t 3 NequIP ,[12] and GemNet,[13] and ra nk as the be st performing models to -da te on test sets, such as the Open Catalyst Project and Direct Air Capture dashboard s. [14][15] GNN -MLIPs do not suer from quadratic memory sca ling with respect to the number of training samples but require signi cantly larger t raining sets to t their large number of pa rameters (tens of thous ands to hun dreds of thousands). GNN model siz e is xed by the GNN architecture , i.e. the number and sizes of GNN embedding s, convolutional, and predictive layers, etc. e sizes of the matrices representing various convolutions and activations re main c onstant across systems as they describe individual elements and atoms. us, GNNs are extendable to any size system and have a c onstant execution cost with respect to tar get size regardless of training set size. However , the modest DFT datasets sizes that are often sucient for constructing a high- performing GAP MLIP are ge nerally insucient to train a GNN MLIP of compara ble accuracy and robustness. erefore, the central diculty in construction of MLIPs is that the model clas s with the most favorable scaling to large problems (GNN-MLIPs) is a lso the one whose data requirements most strongly conict with the cost of DFT data generation. us, improvements to MLIPs for large scale diverse simulations require an improvement in tra ining data collection eciency of GNN-MLIPs. is contribution aims to bridge the knowledge gap of how to achieve high quality GNN training with minimal D F T training data using transfer learning. T ransfer learning is a machine learning technique where a model trained on one task is reused as the initial state for training on another r elated t ask.[16] Neural networks have been demonstrated to ben et from transfer l earning in many diverse tasks, such as imag e classication, [17] machine translation, [18] and 3D pose estimation fr om 2D images,[19] as just a few examples. To -da te, us e of transfer lea rning in atomistic syst ems has be en restri cted to using mo dels pre -trained on large scale g eneralized DFT datasets and th en rened for systems of interest, e.g. Δ- ML .[20] is transfer learning application, therefore, still relie s on the collection of sucient DFT data to fully span th e congurational space of the system of interest. is work demonstrates a novel transfer learning para digm for achieving high quality GNN -MLIPs with minimal DFT training dat a by using classical force elds, which w e call T ransfer-Pa iNN, or T -PaiNN , because we employ the P aiNN ML architecture. Specically , we improve the DFT data eciency of GNN-MLIPs by pre training a PaiNN model on a lar ge dataset (>100 times the DFT data set) generated from classical force eld simulation of the system of interest and then tuning the model weights using the small DFT data set. Classical force elds provide a computationally inexpensive approxima tion of the DFT potential ener gy surface (PES ) and are directly usable in molecular dynamics simulations for generating the large datasets necessary to tra in robust GNN-MLIP . Beca use classical force eld simulations are eectively free in terms of computational time compared to DFT simulations, the computational c ost of this extra step is minimal. W e nd that our T -PaiNN methodology not only signican tly decrease s the DFT data required to train high performance ML IPs, but it also impr oves the generality of the MLIPs performance. Pelletier et al, 20 26 ArXive Preprin t 4 2. Methods is section details the fundamental concept used in T -PaiNN methodology , the underlying graphical network architecture, the exemplar systems tested, and th e computational data collection methods, including both the classical and quantum calculation methods. 2.1 T ransfer learning for Graph Neural Network Prediction of Atomist ic Systems. e c entral hypothesis unde rlying the T - PaiNN workow is that there exists a strong correlation betwee n atomic geometries and the associated ener gy/force data set calculated at both the classical a nd quantum levels because th ey are both methods of modeling the behavior of atomic systems, albeit with varying levels of a ccuracy and cost. erefore, the information embedded into ML IP models trained on classical force elds are directly trans latable to quantum mechanical tasks. is premise ca n be placed into a statistical framing as follows. Given two data sets, a classical forc e eld data set , where is a feature vector representing atom ic structures drawn from sampling distribution with a s the corresponding e ner gy/force label from the target space calculated by classical force elds, and a quantum data se t, , consist ing of labeled as calculated at the quantu m level, if a model can map , only mi nimal information about is nece ssary to re train to mapping . e application of the T -PaiNN workow progresses in three main stages, as shown schematically in Figure 1: 1) the generation o f a classical force eld dataset on the syst em o f interest, 2) pretraining the GNN by the classical forceeld data set, then 3) the transfer learning netuning of the GNN using DFT results. W e refer to this last stage as Autotuni ng T -PaiNN because it tunes the T -PaiNN model weights based on the DFT inform ation used to align the classical and DFT info rmation, and thus this information is automatically provided. e classical mechanics derived pretraining dataset represents a more complete sampling of an approximate PES than is possible using DFT , while the DFT dataset represents a sparse Figure 1 : Schematic of the T-PaiNN procedure, with shapes representing each object’s type and colors representing the quality of the PES for that object. Pelletier et al, 20 26 ArXive Preprin t 5 sampling of a more accurate P ES. e pretraining stage takes advantage of thi s broader sampling by plac ing the model’ s weights into a re gion of parameter spa ce which is consistent with a smooth and physically plausible PES. W ith the weights initialized in this state, autotuning T -PaiNN on the DFT data adjusts the weights to be consistent with the DFT PES without leaving this smooth-PES region of the spa ce. W ithout pre-training and autotuning, DFT -only tting must discover both the or ganization of congura tion space and the mapping from the embedding to the DFT PES from a much more limited set of data. On subsequent ev aluations whe re extrapolations are unavoidable, such as when performing molecular dynamics simulations, the T -PaiNN models are hypothesized to remain more robust beca use they were trained on a far greater number of atomic congurations than equivalent models only trained on DFT da ta (i.e. DFT -only PaiNN models). W e note two im portant points necessary to maximize the performance of T -PaiNN. Firstly , after the classical dataset has been generated, the force eld and DFT potential ener gy surfaces (which are dened only to an additive constant) must be aligned using paired force eld and DFT evaluations on a shared set of structures. is alignment is done without violating any derived quantities (forces, stresses) via lea rned per-atom energy osets for each e lement type derived from the residuals between force-eld and DFT ener gies, and thus is essentially computationally free. Second, a classical for ce eld which is appropriate for the system of intere st should be selected to generate the pretraining data. It is preferable to uti lize an existing force eld that is applicable to the system of intere st whenever possi ble, since force elds that have already been v alidated for molecular dynamic simulations of the systems of interest are more likely to remain numerically robust over the trajectories needed for dataset generation and are li kely to b etter rep resent the P ES. If such a classical fo rce eld does not exist, howev er , tting standard classic al force eld functional forms to the available DFT data is also possib le. Even a relatively simple tted force eld may still provide an informative a pproximation to the DFT potential ener gy surface due to the physical structure imposed by the parameterized functional forms. 2.2 Choice of GNN MLIP Architecture: Polarizable Atom Interaction Neural Network e Polariz able Atom Interaction N eural Network (PaiNN) implementation of GNN-ML IP architecture is used to pre dict ener gies, forces and s tresses from nuclear posi tions and is the chosen architecture of thi s work . However , we note that the transfer app roach is agnostic to architecture and is generally applicable to all GNN-MLIPs. PaiNN implements a Deep S ets approach;[21] specically , it embeds each atom into a latent representation space, p erforms interaction operations between atoms within this space according to the graph connectivity of the atoms, and performs nal inference by collapsing these atomic e mbeddings into the quantity o f interest, in our case, system energy . Interactions between atoms in the latent space are executed using a message passing GNN. Massage passing GNN’ s provide enough complexity to perform favorably on benchmark datasets[15] while remaining modest in parameter size, thus making its training more feasible. In this work, we constr uct the PaiNN architecture with the hyperparameters re ported in T able 1. W e note that some models available online exceed these parameter counts by a factor of Pelletier et al, 20 26 ArXive Preprin t 6 10,; however , because the purpose of this contribution is focused on gains enabled by infor mation transfer rather than maximizing performance, we use the more modest PaiNN size to avoid the additional computational cost of lar ger model training. Table 1 : PaiNN Hyperparameters for the 3 model sizes used in this study. Small Model Medium Model Large Model Atomic Embedding Dimension 32 64 128 Interaction Updates 3 4 5 Radial Basis Functions 16 32 32 Parameter Count 36,300 197,000 534,000 2.3 T -PaiNN T est S ystems T wo test systems were selected to demonstrate the performa nce of T -PaiNN method: 1) gas phase molecular syste ms taken from the QM9 da tabase,[22] and 2) liquid phase bulk water calculated in this wor k. ese systems were chosen to r epresent major classes of chemica l systems examined in atomistic modeling while maintaining computational tractability by using the extant QM9 dataset and limiting the number of atom types in the other system. Each of these systems is d iscussed further below , including the computational parameters used for the models. 2.3.1 Gas Phase Molecular Systems – e QM9 Data Set Gas phase molecular systems were extracted fro m the QM9 molecular energy database. [22] e QM9 databa se compiles ~134,000 molecular structures containing the eleme nts H, C, N, O, and F and their ener gies as calculated by an array of dierent accuracy quantum chemical methods. W e chose to use the QM9 D FT data calculated at the B3L YP exchange correlation functional level of DFT with a 6-31G(2df,p) basis set.[23][24] e uni versal for ce eld (UFF)[25] classical force eld was used to calculate the energies of these molecules in the LAMMPS prog ram and was used for pretraining the T -PaiNN model.[26] Given the large number of structures, only single point ener gies w ere collected from the QM9 tabulated geometries; no geometry optimizations or molecular dynamics runs were used to gather a lar ge data set. e accuracy mea sure of the various models on the QM9 dataset was selected to be the molecular ener gy of th e system. 2.3.2 Liquid Phase System – Bulk W ater Data Set Bulk liquid phase water wa s selected as a repre sentative for liquid syste ms because of its ubiquity in c hemistry and biology , and its complex behavior relative to its small size, which arises from the complex combined ionic a nd covalent character of its hydrogen bonding network. e TIP3P water force eld served as the classical forceeld for pretraining T -PaiNN.[27] While this for ce eld is Pelletier et al, 20 26 ArXive Preprin t 7 typically used with a rigid constraint plac ed on the water molecules, which prohibits O- H vibrations and H-O-H an gle extensions, a exible version of thi s force el d was chosen to b etter align the results with the DFT tar get where the angle and bond lengths are unconstrained. e TIP3P force eld was used within LAMMPS to run an NPT MD trajectory of 520 waters for 1,000,000 steps of 0.25 fs each, with eve ry 1,000th frame extr acted to c onstruct the pretraining dataset. A DFT dataset was generated from two sepa rate AIMD trajectories performed in V ASP with the SCAN functional using a simila r procedure, resulting in 879 structures being extracted in total, 839 of which contain 65 waters and 40 of which contai n 136 waters. [28][29] e SCAN meta-GGA functional was chosen due to its comparatively strong performance on liquid water wh en c ompared to other GGA or meta-GGA fun ctionals.[30] Plane waves with ener gies up to 700 eV were used to construct the wavefunctions. e Brill ouin zone was only sampled at the Γ-point owing to the la rge size, disordered struc ture, and insulating nature of the water molecules. As with the QM9 case, t hree DFT -only training sets of dierent sizes (50, 100, and 500) were created by random selection to test the data eciency improvements aorded by the T -PaiNN procedure. e remaining structures were used for hold -out testing. e accuracy of the va rious models on the water sy stem wa s eva luated by comparing their e nergies, atomic forc es and calculated pressures in addition to properties commonly extrac ted from molec ular dynamics simulations, particularly predicted density , radial distribution function s, and water diusivity , for which experimental values are well documented. 2.4 Code Development and Packages All code was developed and written in python and MLIP construction and training was c onducted using pytorch.[31] e SchNe tP ack python code w as used for generating an d executing the PaiNN MLIP architecture .[32] e Atomic Simulation Environment (ASE) package was used for running the MD simulation using the T -PaiNN derived MLIPs.[33] 3. Results and Discussion T o determine the viability of the T -PaiNN method, we benchmar ked the transfe r leaning capabilities on two systems: the QM9 benchmark small mol ecule dataset and simulating liquid water . e former demonstrates pe rformance across a wide va riety of molecular systems while the later demonstrates performance in complex inter-molecular systems. In all cases, the only input into the machine learned networks are the atomic positions, and the outputs are energies for the QM9 models a nd energies, forces, and stresses for the water models. From these, all other quantities are derived. 3.1 Small Molecule Energies W e examined the use of the T -PaiNN approach to non-interacting, i.e. ga s pha se, molecular systems through the QM9 dataset. e QM9 data was randomly partitioned into three se ts: Pelletier et al, 20 26 ArXive Preprin t 8 pretraining, validation and test. A subset of 50,000 struc tures from the 134,000 in the QM9 database were randomly selected to for m the pret raining set for calculation at the UFF level , as shown in F igure 2.a. is number was selected to ensure heterogeneity of molecules and syst ems without the need for calculating molecular dynamic trajectories. e validation set, used to detect overtting and trigger early stopping of th e training , comprised 20,000 molecules. e test set contained the remaining ~60,000 molecules. Figure 2: (a) A visualization of how the QM9 dataset was divi ded to form the sets used in this task, along with some randomly chosen example molecules from the dataset. (b) e energy residuals between the p reexisting UFF force eld with atomic energy osets added and the corresponding DFT energies for all molecules in the QM9 dataset. 3.1.1 Generation of Pr etraining Set and Alignment of Ener gies T o align the classical force eld and DFT poten tial ener gy surfaces, e lemental atomic energy osets were calculated via linear regression between molecule elem ent counts and their UFF to DFT e ner gy residuals. is was performed using only molecules in the DFT tra ining sets, of sizes 100, 500 or 2,500 depending on the DFT training set used for autotuning. e residual distribution of the UFF e ner gies aligned to 500 DFT examples is shown in Figure 2.b. e mean absolute e rror of 6.9 eV is a 57% improvement from the atomic-o set-only MAE of 12.2 eV (i.e. what is exp ected just base d on the number of ea ch element a nd their ene r gies). is improvement demonstrat es that the classical force eld p artially capture s the phys ics of the molecul es ; however , the 6.9 eV error is insucient for examining detailed chemistry . e error distribution is bimodal ; the negative mode peaks near -5.16 eV and predominantly comprises pure hydrocarbons while the posi tive mode peaks near +4.85 e V and predominantly contains N and O-rich mol ecules. ese large and systematic errors indicate that the force eld is doing a poor job c haracterizing the detailed chemistry of these molecules, despite getting some of the behavior correct. Ne vertheless, these ener gies provide basic information whic h is su cient to serve as the initial T -PaiNN model training set. (b) (a) Pelletier et al, 20 26 ArXive Preprin t 9 3.1.2 Model Accuracy and T raining Speed of T -Pa iNN on Molecular Systems T o test the improvement in data ecienc y aorded by the T -PaiNN procedure, three small PaiNN models (36.3k parameters) were generated and trained on the 100, 500, and 2,500 molecule datasets to serve as DFT -only models. ese models are ref erred to by the number of exemplar DFT molecules included. An identical PaiNN model (36.3k pa rameters) was tra ined on the 50,000 UFF dataset and served a s the standard initial state for T -PaiNN models, whic h was autotuned on each of the three DFT datasets. is procedure resulted in a total of seven PaiNN models: three DFT -only models trained only on varying amounts DFT da ta, one model based on the UFF only dataset, and three T -PaiNN models trained on the 50,000 UFF dataset and then autotuned on the three DFT training sets. As these models trained, they were evaluated on a completely held-out test set of 60,000 molecules from the QM9 d ataset, the resulting training loss curves for both the DFT -only PaiNN models and the T -PaiNN models are shown in Figure 3. Figure 3 : QM9 test-set p erformance for the DFT -only (black) and T-PaiNN (blue) GNN MLIPs over the course of their training. e curves vary in the number of epochs due to validation set triggered early-stopping during training, which is indicated by a star at the end of the curve. For a ll tra ining set sizes, the T -PaiNN models outperform the DFT -only models in terms of both global minimum MAE on the test set and the number o f DFT training epochs required to achieve optimal perform ance. e DFT -only P aiNN models achieved mi nimum test set MAE values of 16.2, 4.5, and 0.8 eV from the 100, 500, and 2,500 DFT training sets respec tively , while T -PaiNN models ac hieved minimum MAE values of 0.6, 0.8, a nd 0.5 eV wh en trained on the same DFT training sets. ese correspond to 25.3 , 5.9 , and 1.6 reductions in MAE for the same amount of expensive DFT calculations. In addition to this dramatically improved performance, the T -PaiNN models reached their accuracy-plateau more quick ly than the DFT -only models. Model training speed-up is quantied by nding the rst epoch whe re test MAE is within 10% of the minimum value. e T -PaiNN method showed a 5.7 , 3. 5 , and 2.0 speedup on the 100, 500, and 2,500 sized training sets, achieving convergence in 9 , 14, and 31 epo chs respectively compared to 51, 49, and 62 for the (b) (c) (a) Pelletier et al, 20 26 ArXive Preprin t 10 DFT -only models. us, the T -PaiNN method not only increases accuracy , but achieves conver g ence faster than the simple DFT only method. 3.1.3 Model Size Eects of T -PaiNN e T -PaiNN procedure performance was evaluated as a function of model size with medium and lar ge size d PaiNN models (197k and 534k para meters, respectively as shown in T able 1). is test examined just the 500 DFT dataset for both T -PaiNN and the DFT only P aiNN models . e training ac curacy results are shown in Figure 4 and are similar to those found with the smaller models, but with lower error for both the T -PaiNN and DFT only systems . e DFT -only model reached a minimum test set MAE of 1. 45 eV on epoch 59, while the T -PaiNN model reac hed a minimum of 0.23 eV on epoch 98, a 6.3 improvement. It is crucial to note that with the 0.23 e V error m ark, T -PaiNN is approaching expected B 3L YP accuracy to the true chemical energies ; conversely , the 1.45 eV MAE of the DFT only PaiNN model renders it useless for examining chemistry . Across the 100 training epochs, only thr ee individual T -PaiNN epo chs (at the vary beginning) had test set MAEs worse than the best DFT -only model at its end. Additionally , fr om epochs 50 to 100 (chosen to capture the accuracy platea us fo r both models), the T -PaiNN model demonstrated much gr eater stability than the DFT -only model, with a standard deviation of 0.4 eV in MAE compared to 3.2 eV . ese re sults indi cate both improved performance and st ability of the T -PaiNN models compared to DFT -only training. All results for the molecular systems are aggregated in T able 2. Overall, the T -PaiNN method greatly increases the accuracy of GNN -MLIPs at minimal extra cost of data collection, while simult aneously smoothing epoch- to -epoch training variance and decreasing time to DFT tra ining conver gen ce in molecular systems. Figure 4 : QM9 test-set performance for DFT -only (black) and T-PaiNN (blue) PaiNN models of (a) small (36k parameters), (b) medium (197k paramete rs), and (c ) lar ge (534k parameters), as they were trained on the 500 dataset. e curves vary in the number of epochs due to early stopping being used during training based on the validation data, which is indicated by a star at the end of the curve. (b) (c) (a) Pelletier et al, 20 26 ArXive Preprin t 11 Table 2 : Summary of best performance achieved on the QM9 test set for eac h model. DFT Training Set Size Model Parameters Minimum MAE (eV ) Epochs to Converg ence DFT-only T-PaiNN Improvement DFT-only T-PaiNN Improvement 100 36,300 16.2 0.6 25.3 51 9 5.7 500 36,300 4.49 0.76 5.9 49 14 3.5 2,500 36,300 0.84 0.52 1.6 62 31 2.0 500 197,000 1.13 0.32 3.5 92 67 1.6 500 534,000 1.60 0.23 6.3 77 63 1.2 3.2 Liquid Water Beha vior A major hurdle in exa mining aqueous chemistry i s the representation of solvated systems . T o do so has conventionally required trade-o b etween low -cost, lower -accuracy implicit solvent methods and higher -cost , higher-accuracy explicit solvation. MLIPs are highly promisi ng for explicitly represent solva ting systems while retaining quantum leve l accuracy . However , gathering sucient quantum data across vast conguration space associated with liquids with which to construct the MLIP is daunting. W e aim to examine the ability of T -PaiNN to overcome this challenge using water as an exemplar system. For this sec tion, only the lar ge PaiNN ar chitecture was used, see T able 1 for hyperparameters. Unlike the QM9 task, which required only the prediction of molecular ener gies, th is task examines the generation of a model that can pe rform molecular dynamics runs of water , and therefore high acc uracy ener gies, forces, and sim ulation ce ll stresses are all require d. Forces and stresses are both properties derived from dierentiating the potential energy function of the system with respect to a tomic coordinates or cell lattice parameters, respectively , both of whic h can e asily be obtained from a GNN-MLIP by autodierentiation. 3.2.1 Alignment of Classical and Quantum V alues for Liquid W ater System e TI P3P force eld be havior correlates well with the S CAN DFT calculations, as shown by the correlation between the predicted energy , force ma gnitude, and force dir ection by the two methods, shown in F igure 5. V irial stress values (aggregate d into pressures) show considerable correlation ( ), but a lar ge MAE of 2376 bar . is disagreement arises from the known bias of TIP3P to genera lly under -predict the density of liquid water and the tendency of SCAN-DFT to over- predict the density . However , th e underlying correlation is sucient to translate the classical ener gies, forces and stresses into the DFT e ner gy scale. Pelletier et al, 20 26 ArXive Preprin t 12 Figure 5: Comparison of TIP3P ene rgies, for ces, and stresses (a ggregated into pressures) to those of DFT on the same structures. (a) Shows th e parity plot between atomic energy adjusted TIP3P energies and DFT energies, (b) shows the parity of the pressures o f these structures, (c) shows a histogram of force magnitude errors, and (d) shows a histogram of force vector alignment errors. 3.2.2 Liquid W ater T -PaiNN Performance with V arying Amounts of DFT Data e training acc uracy of the dierent models are shown in F igure 6. Simil ar ly to the QM9 task, the T -PaiNN models demonstrate im proved performance ac ross the boa rd. In terms of ener getics (Figure 6.a-c) the DFT -only models showed minimum test set MAE values of 0.49, 0.46, and 0.39 eV from the 50, 100, 500 datasets respectively , while the T -PaiNN models had minimum test set MAEs of 0.33, 0.30 and 0.27 eV . From epochs 100 to 200 (chosen to represent the accuracy plateau re gion), the T -PaiNN models de monstrated improved stability , wi th standard deviations in test set MAE of 0.32, 0.36, and 0.27 eV , in comparison to 1.89, 1.95, and 0.95 for the DFT -only models. us, in addition to the improvements in MAE, the T -PaiNN mod els appear to tr ain within a smoother and more sta ble region of parameter space . is is particularly encouraging for the extrapolation abilit y of T -PaiNN trained models, and the stability of long molecular dynamics runs. W e hypothesize that the signi cant, but lower factional, improvement of T -PaiNN on the wa ter system compare d to the QM9 system is that water is the only molecule which nee ded to be lea rned by the MLIP . Pelletier et al, 20 26 ArXive Preprin t 13 Figure 6 : DFT water test set performance for the DFT-only (black) and T-PaiNN (blue ) models during their training on three dierent sized DFT training sets, 50 (a,d,h) , 100 (b,e,i) , and 500 (c,f,j) . e rst row (a,b,c) shows energy MAE, the second (d,e,f) shows force vector component MAE, and th e third (h,i,j) shows stress component MAE. For the energy subplots, the rst 50 e pochs are shown in the main plots, highlighting the early training improvements, with all 200 epochs shown in the inset. T -PaiNN models demonstrate ev en stronge r im provements in terms of ca lculated for ce as shown by the MAEs in F igure 6.d-f. e three DFT -only models reached minimum MAEs of 0.12, 0.1 1, and 0.09 when trained on the 50, 100, 500 datasets respectively , while the corresponding T -PaiNN models were approximately half that, reaching minim um MAEs of 0.06 , (a) (b) (c) (d) (e) (f) (g) (h) (i) Pelletier et al, 20 26 ArXive Preprin t 14 0.05, and 0.04 . For all T -PaiNN models, the force MAEs continued to improve until the end of the 200 epoch training, with the minima found at epochs 199, 197 and 198 for the 50, 100, and 500 models respectively . is was also true for the 50 DFT -only model, but for the other two th e fo rce MA Es stagnated or b egan t o increase for th e 100 and 500 DFT -only models respectively , indicating overtting which the corresponding T -PaiNN models did not experience. e stress MAE training curves shown in Figure 6.h-j show similar behavior to the force MAE curves. e three DFT -only models demonstrated MAE minima of 2.1, 2.0, and 1.8 10 3 for the 50, 100, and 50 0 models, with the corresponding T -PaiNN models demonstrating minima of 1.8, 1.7, and 1.7 10 3 . In this case, all three DFT -only models either stagnated or began to degrade in stress p erformance over the cou rse of the 200 epoch training. e thre e T -PaiNN models conversely do not demonstrate any degradation of performance, with the 50 model stagnating, but the 100 and 500 T -PaiNN models demonstrate continued improvement, with their minima occurring at epo chs 186 and 197 out of 200. 3 .2.2 Molecular Dynamics Simulation of Water using T-PaiNN and DFT-only PaiNN MLIP While the liquid water results of Section 3.2 suggest that the T -PaiNN procedure improves the prediction of DFT wa ter ene r gies, forces, and stresses, a more im portant qu estion is whe ther these gains translate into e nhanced physical prediction s in molec ular dynamics simulation. Molecular dynamics simulation of liquid water ther efore provides a more releva nt test of the T -PaiNN methodology than merely ho ld -out predicted energy and force error alone. e performance of three ML force eld models, a DFT -only PaiNN, a TI P3P-only PaiNN (pretrained), and T -PaiNN, are examined for accurac y across three exp erimental observa bles: water de nsity , the water radial distribution functions, and water self-diusion. e results are shown in Figure 7. Figure 7 : e performance of the DFT-only PaiNN, TIP3P -only P aiNN, and T -PaiNN models in 50ps NPT MD simulations of a box of 520 waters. (a) e density of the water box for the thre e water models, (b) their radial distribution functions, (c) the mean square displacement of the oxygen atoms over the simulations. (a) (b) (c) Pelletier et al, 20 26 ArXive Preprin t 15 Across all three experimental observa bles, the T -PaiNN MLIP properties match closer to experiment than the other two PaiNN models, as tabulated in T able 3. e PaiNN model derived from the classical force eld pre-training unde restimates the density while overestimating the hydration shell structuredness (higher pe ak around 3Å in the RDF of Figure 7.b), and self- diusivity of water . e DFT -only trained PaiN N model overestimates the water destiny a nd underestimates the structural rigidity and self-diusion. ese are known limitations of the TI P3P and SC AN-DFT water PESs. T -PaiNN is closer to experiment than the other two across all metrics aside from four (see T able 3) : the int ensities of the RDF peaks as calculated by DFT only PaiNN, which performs worse than T -PaiNN in predicting peak location, and th e rst peak location of TIP3P . Given the importanc e of p eak location, we posit that a <10% error in peak intensity compared to experiment and DFT is acceptable, gi ven the very g ood agreement with peak location. Beyond being the model which best reproduces ex periment, the T -PaiNN model is more similar to the DFT -only model than the pretrained PaiNN model . us, we conclude th at the transfer learning processes embeds the model with the key energetic information from DFT and that the improved performance compared to DFT -only arises f ro m being exposed to a larger number of congurations. Table 3 : Comparison of DFT-only and T-PaiNN models performance on the liquid water task. Metric DFT-only TIP3P-only T-PaiNN Experiment Improvement Intrinsic † Energy ( MAE ) 0.36 1.08 0.26 - 1.40 Force ( MAE) 0.09 0.23 0.03 - 2.80 Stress ( MAE) 0.0018 0.0023 0.0017 - 1.05 Derived Density ( ) 1.07 0.94 1.02 0.99 - Self-Diffusion ( 10 5 ) 1.50 4.08 1.87 2.30 - RDF Extrema 1 st Peak Position 2.78 2.78 2.78 2.80 - Intensity 2.34 2.95 2.73 2.58 - 1 st Trough Position 3.28 3.33 3.28 3.47 - Intensity 0.84 0.82 0.75 0.84 - 2 nd Peak Position 3.98 4.38 4.48 4.51 - Intensity 1.13 1.03 1.20 1.12 - 2 nd Trough Position 5.48 4.48 5.58 5.58 - Intensity 0.86 1.04 0.84 0.88 - 3 rd Peak Position 6.58 6.78 6.73 6.69 - Intensity 1.09 1.03 1.07 1.06 - Wins 3 1 10 † Comparison to SCAN-DFT evaluated held-out test set. ‡ Derived from 50ps NPT molecular dynamics simulation of 520 waters. Pelletier et al, 20 26 ArXive Preprin t 16 4. Conclusions Herein, we have developed and demonstrated a novel T ransfer-PaiNN method for tr aining interatomic graph neural network potentials. By pretraining network with low -cost, low-accuracy classical force led data, and subsequently autotuning the resulting model with DFT data, the resulting autotuned T -PaiNN model signicantly and persistently outperforms models trained on DFT data alone. e im proved performance holds across mol ecular and periodic boundary condition syste ms a nd systems with intra- and inter- mo lecular for ces. W e nd performance improvements of 2-10 , and that this method brings the error closer to th e DFT expected accuracy . W e hypothesize that this perf ormance improvement arise s from the model learning general interatomic intera ctions from a much lar ger range of systems. us, the mode l is less likely to rely on extrapolation than DFT only models. Overall, these results show that T -PaiNN is a practical methodology for signicantly improving graphical neural ne tworks for atomic calculations with minimal additional computational cost. 5. Fund ing Sources e Author s gratefully acknowledge seve ral funding sources which supported the authors contributing to thi s cross-project collaborative eort. VP acknowledges support from the Science and T e chnologies fo r Ph osphorus Sustainability (STEPS) Center , a Nation al Science Foundation Science and T echnology Center (CBET -2019435). VB acknowledges support from U.S. Department of Energy , Oce of Science, Oce of Basic Ener gy Sciences , under A ward Number DE -SC0024194. CM and DR acknowledge support from the U.S . Na tional Science Founda tion CBET Catalysis under award 2450869. SR acknowledge s support from the DOE Oc e o f Science, Oce o f Basic Energy Sciences (BES), Materials S ciences and Engineering Division under A ward DE-SC0024724. e content is solely the a uthors’ responsibility and does not necessarily repre sent the ocial views of the National Institutes of Health, US Department of Ene rgy , or the NSF . In addition, we a cknowledge support from Research Computing at Arizona State University for providing high-performance supercomputing services. 6. References [1] R. Jacobs et al. , “A practical guide to machine learning interatomic potentials – Status and future,” Curr . Opin. Solid State Mater . Sci. , vol. 35, p. 101214, Mar . 2025, doi: 10.1016/j.cossms.2025.101214. Pelletier et al, 20 26 ArXive Preprin t 17 [2] A. P . Bartók, M. C. Payne, R. Kondor , and G. Csá nyi, “Gaussian Approximation Potentials: e Accuracy of Quantum Mechanics, without the Electrons,” Phys. Rev . Lett. , vol. 104, no. 13, p. 136403, Apr . 2010, doi: 10.1 103/PhysRevLett.104.136403. [3] J. Behler and M. Parrinello, “Generalize d Neural- Network Representation of High- Dimensional Potential-Ener gy Surfaces,” Phys. Rev . Lett. , vol. 98, no. 14, p. 146401, Apr . 2007, doi: 10.1 103/PhysRevLett.98.146401. [4] Y . Y ang, O. A. Jimenez-Negron, and J. R. Kitchin, “Machine-learning accelerated geometry optimization in molecular simulation,” Apr . 2021, doi: 10.1063/5.0049665. [5] J. A. Garrido T orres, P . C. Jennings, M. H. Hansen, J. R. Boes, and T . Bligaard, “Low- Scaling Algorithm for Nudged Elastic Band Calculations Using a Surrogate Machine Learning Model,” Phys. Rev . Lett. , vol. 122, no. 15, p. 156001, Apr . 2019, doi: 10.1 103/PhysRevLett.122.156001. [6] B. Deng et al. , “CHGNet as a pretrained universal neural network potential for char ge - informed atomistic modelling,” Nat. Mach. Intell. , vol. 5, no. 9, pp. 1031–1041, Sep. 2023, doi: 10.1038/s42256-023-00716-3. [7] R. Jinnouchi, F . Karsai, and G. Kresse, “Making free-ener gy calculations routine: Combining rst principles with machine learning,” Phys. Rev . B , vol. 101, no. 6, p. 060201, Feb. 2020, doi: 10.1 103/PhysRevB.101.060201. [8] V . L. Deringer , A. P . Bartók, N. Bernstein, D. M. W ilkins, M. Ceriotti, and G. Csányi, “Gaussian Process Regression for Materials a nd Molecules,” Chem. Rev . , vol. 121, no. 16, pp. 10073–10141, Aug. 2 021, doi: 10.1021/acs.chemrev .1c00022. [9] C. E. Rasmussen and C. K. I. W illiams, Gaussian Pr ocesses for Machine Learning . e MIT Press, 2005. doi: 10.7551/mitpress/3206.001.0001. [10] K. T . Schütt, P .-J. Kindermans, H. E. Sauceda, S. Chmiela, A. Tka tchenko, and K.-R. Müller , “SchNet: A conti nuous-lter convolutional neural network for modeling quantum interactions,” Dec. 2017. [1 1] K. T . Schütt, O. T . Unke, and M. Gastegger , “Equivariant message passing for the prediction of tensorial properties and molec ular spectra,” Jun. 2021. [12] S. Batzner et al. , “E(3)-equivariant graph neural networks for data-ecient and accurate interatomic potentials,” Nat. Commun. , vol. 13, no. 1, p. 2453, May 2022, doi: 10.1038/s41467-022-29939-5. Pelletier et al, 20 26 ArXive Preprin t 18 [13] J. Gasteiger , F . Becker , and S. Günnemann, “GemNet: Universal Directional Graph Neural Networks for Molecules,” Jun. 2024. [14] L. Chanussot et al. , “Open Catalyst 2020 (OC20) Dataset and Community Challenges,” ACS Catal. , vol. 1 1, no. 10, pp. 6059–6072, May 2021, doi: 10.1021/ac scatal.0c04525. [15] A. Sriram et al. , “e Open DAC 2023 Dataset and Challenges for Sorbent Discovery in Direct Air Capture,” ACS Cent. Sci. , vol. 10, no. 5, pp. 923 –941, May 2024, doi: 10.1021/acscentsci.3c01629. [16] A. Hosna, E. Merry , J. Gyalmo, Z. Alom, Z. Aung, and M. A. Azim, “T ransfer learning: a friendly introduction,” J. Big Data , vol. 9, no. 1, p. 102, Oct. 2022, doi: 10.1 186/s40537- 022-00652- w. [17] M. Long, Y . Cao, J. W ang, and M. I. Jordan, “Lea rning T ransferable Features with Deep Adaptation Networks,” May 2015. [18] B. Zoph, D. Y uret, J. May , and K. Knight, “T ransfer Learning for Low-Resource Neural Machine T ranslation,” in Pr oceedings of the 2016 Confer ence on Empirical Methods in Natural Language Pr ocessing , Stroudsburg, P A, USA: Associ ation for Computational Linguistics, 2016, pp. 1568–1575. doi: 10.18653/v1/D16-1 163. [19] D. Mehta et al. , “Monocular 3D Human Pose Estimation In e W ild Using Improved CNN Supervision,” Oct. 2017. [20] X. Chen, P . Li, E. Hruska, and F . Liu, “∆-Machine Learning for Quantum Chemistry Prediction of Solution-phase Molecular Properties at the Ground and Excited States,” Feb. 03, 2023. doi: 10.26434/chemrxiv-2023-ddcr1. [21] M. Zaheer , S. Kottur , S. R avanbakhsh, B. Poczos, R. Salakhutdinov , and A. Smola, “Deep Sets,” Apr . 2018. [22] S. Nandi, T . V egge, and A. Bhowmik, “MultiXC-QM9: Large dataset of molecular a nd reaction ener gi es from multi -level quantum chemical methods,” Sci. Data , vol. 10, no. 1, p. 783, Nov . 2023, doi: 10.1038/s41597-023-02690-2. [23] A. D. Becke, “Density-functional thermochemistry . III. e role of exact exchange,” J. Chem. Phys. , vol. 98, no. 7, pp. 5648–5652, Apr . 1 993, doi: 10.1063/1.464913. [24] C. Lee, W . Y ang, and R. G. Parr , “Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density ,” Phys. Rev . B , vol. 37, no. 2, pp. 785– 789, Jan. 1988, doi: 10.1 103/PhysRevB.37.785. Pelletier et al, 20 26 ArXive Preprin t 19 [25] A. K. Rappe, C. J. Casewit, K. S. Colwell, W . A. Goddard, and W . M. Ski, “UFF , a full periodic table force eld for molecular mechanics and molecular dynamics simulations,” J. Am. Chem. Soc. , vol. 1 14, no. 25, pp. 10024–10035, Dec. 1992, doi: 10.1021/ja00051a040. [26] A. P . ompson et al. , “LAMMPS - a exible simulation tool for partic le-based materials modeling at the atomic, meso, and continuum scales,” Comput. Phys. Commun. , vol. 271, p. 108171, Feb. 2022, doi: 10.1016/j.cpc.2021.108171. [27] W . L. Jorgense n, J. Chandrasekhar , J. D. Madura, R. W . Impey , and M. L. Klein, “Comparison of simple potential functions for simulating liquid water ,” J. Chem. Phys. , vol. 79, no. 2, pp. 926–935, Jul. 1983, doi: 10.1063/1.445869. [28] G. Kresse and J. Hafner , “Ab initio molecular dynamics for liquid metals,” Phys. Rev . B , vol. 47, no. 1, pp. 558–561, Jan. 1993, doi: 10.1 103/PhysR evB.47.558. [29] J. Sun, A. Ruzsinszky , and J. P . Perdew , “Strongly Constrained and Appropr iately Normed Semilocal Density Functional,” Phys. Rev . Lett. , vol. 1 15, no. 3, p. 036402, Jul. 2015, doi: 10.1 103/PhysRevLett.1 15.036402. [30] R. W ang, V . Carnevale, M. L. Klein, and E. Borguet, “First-Principles Calculation of W ater pKa Using the Newly Developed SCAN F unctional,” J. Phys. Chem. Lett. , vol. 1 1, no. 1, pp. 54–59, Jan. 2020, doi: 10.1021/acs.jpclett.9b02913. [31] A. Paszke et al. , “PyT or ch: An Imperative Style, High-Performance Deep Learning Library ,” Dec. 2019. [32] K. T . Schütt, P . Kessel, M. Gastegger , K. A. Ni coli, A. Tka tchenko, and K.-R. Müller , “SchNetPack: A Deep Learning T oolbox F or Atom istic Systems,” J. Chem. eory Comput. , vol. 15, no. 1, pp. 448–455, Jan. 2019, doi: 10.1021/acs.jctc.8b00908. [33] A. Hjorth Larsen et al. , “e atomic simulation environment—a Python library for working with atoms,” Journal of Physics: Condensed Matter , vol. 29, no. 27, p. 273002, Jul. 2017, doi: 10.1088/1361-648X/aa680e.
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment