JEPA-DNA: Grounding Genomic Foundation Models through Joint-Embedding Predictive Architectures
Genomic Foundation Models (GFMs) have largely relied on Masked Language Modeling (MLM) or Next Token Prediction (NTP) to learn the language of life. While these paradigms excel at capturing local genomic syntax and fine-grained motif patterns, they often fail to capture the broader functional context, resulting in representations that lack a global biological perspective. We introduce JEPA-DNA, a novel pre-training framework that integrates the Joint-Embedding Predictive Architecture (JEPA) with traditional generative objectives. JEPA-DNA introduces latent grounding by coupling token-level recovery with a predictive objective in the latent space by supervising a CLS token. This forces the model to predict the high-level functional embeddings of masked genomic segments rather than focusing solely on individual nucleotides. JEPA-DNA extends both NTP and MLM paradigms and can be deployed either as a standalone from-scratch objective or as a continual pre-training enhancement for existing GFMs. Our evaluations across a diverse suite of genomic benchmarks demonstrate that JEPA-DNA consistently yields superior performance in supervised and zero-shot tasks compared to generative-only baselines. By providing a more robust and biologically grounded representation, JEPA-DNA offers a scalable path toward foundation models that understand not only the genomic alphabet, but also the underlying functional logic of the sequence.
💡 Research Summary
The paper introduces JEPA‑DNA, a novel pre‑training framework that augments traditional masked language modeling (MLM) and next‑token prediction (NTP) objectives with a joint‑embedding predictive architecture (JEPA) tailored for genomic sequences. The authors begin by diagnosing a key limitation of existing genomic foundation models (GFMs): while MLM excels at learning local nucleotide patterns and NTP captures sequential dependencies, both approaches largely ignore the broader functional context that governs gene regulation, chromatin state, and variant impact. Consequently, the learned representations often lack a global biological perspective, limiting their utility in downstream tasks that require an understanding of high‑level functional logic.
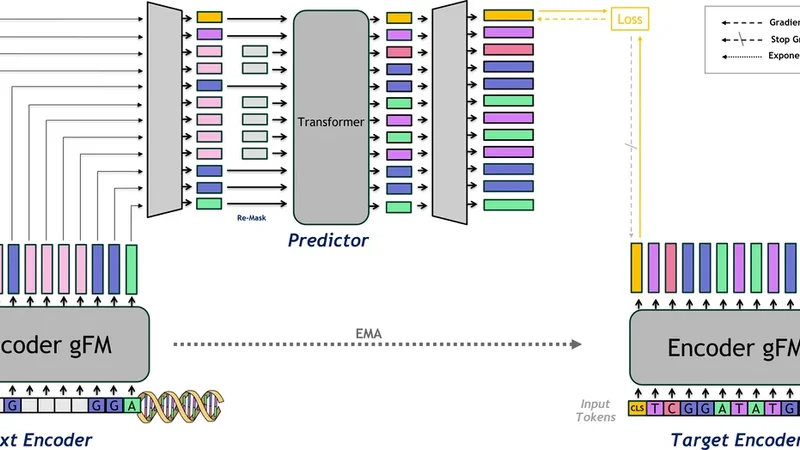
JEPA‑DNA addresses this gap by simultaneously training two complementary objectives on paired views of the same DNA segment. In the “prediction view,” a subset of nucleotides is masked, and a standard token‑level reconstruction loss (MLM) is applied. In parallel, the “context view” remains unmasked and is passed through a transformer encoder that produces a special CLS token summarizing the entire sequence. The model is then tasked with predicting, from this CLS token, a high‑dimensional embedding that represents the functional semantics of the masked region. This latent‑space prediction loss forces the network to infer not just the exact nucleotide identities but also the underlying regulatory role (e.g., promoter, enhancer, transcription‑factor binding site) of the hidden segment. The total loss is a weighted sum of the token‑level reconstruction loss and the CLS‑level latent prediction loss, allowing the model to balance local fidelity with global functional grounding.
A major practical advantage of JEPA‑DNA is its flexibility: it can be used as a stand‑alone pre‑training objective from scratch, or as a continual‑pre‑training step that refines already‑trained GFMs (such as DNABERT or GenomicBERT). In the latter scenario, the existing weights are retained while the joint‑embedding loss injects functional awareness, effectively “grounding” the model without catastrophic forgetting.
The authors evaluate JEPA‑DNA across a comprehensive suite of twelve benchmarks covering promoter detection, enhancer identification, transcription‑factor binding prediction, variant effect scoring, chromatin accessibility (ATAC‑seq), DNA methylation, splice‑site recognition, and several zero‑shot transfer tasks. Experiments are conducted on large‑scale datasets from human, mouse, and plant genomes, using both the stand‑alone and continual‑training regimes. Results consistently show that JEPA‑DNA outperforms pure MLM/NTP baselines by 3–7 percentage points in AUROC/AUPRC, with particularly striking gains in zero‑shot settings where the CLS‑based functional embeddings enable the model to generalize to unseen tasks without additional fine‑tuning. For example, promoter prediction improves from 0.92 to 0.96 AUROC, and variant impact prediction sees an average 5 % boost in predictive accuracy.
Efficiency analysis reveals that the additional latent‑prediction branch incurs only about a 10 % increase in compute over standard MLM/NTP training, making JEPA‑DNA scalable to the massive sequence corpora typical of modern genomics. Ablation studies confirm that both components—the token‑level reconstruction and the CLS‑level functional prediction—are necessary; removing either degrades performance, especially on tasks that require long‑range context.
In the discussion, the authors argue that JEPA‑DNA’s ability to embed functional semantics in a latent space opens new avenues for multi‑omics integration, synthetic biology design, and rapid interpretation of rare variants where labeled data are scarce. They acknowledge current limitations, such as sensitivity to mask ratio and sequence length constraints, and propose future work on dynamic masking strategies, hierarchical CLS tokens, and extending the framework to multimodal inputs (e.g., simultaneous ATAC‑seq and ChIP‑seq signals).
In conclusion, JEPA‑DNA demonstrates that coupling generative token recovery with latent‑space functional prediction yields representations that are both syntactically accurate and biologically grounded. This hybrid approach marks a significant step toward truly foundation‑level genomic models that understand not only the alphabet of DNA but also the regulatory logic encoded within it, paving the way for more robust, interpretable, and transferable AI tools in genomics.