Parameter-free representations outperform single-cell foundation models on downstream benchmarks
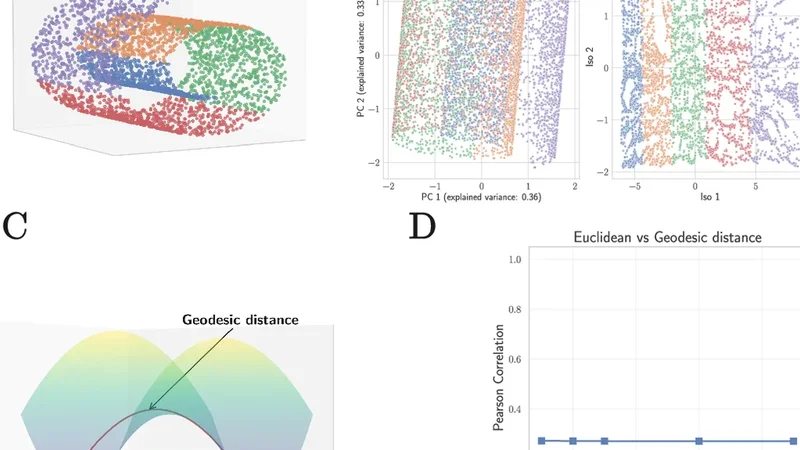
Single-cell RNA sequencing (scRNA-seq) data exhibit strong and reproducible statistical structure. This has motivated the development of large-scale foundation models, such as TranscriptFormer, that use transformer-based architectures to learn a generative model for gene expression by embedding genes into a latent vector space. These embeddings have been used to obtain state-of-the-art (SOTA) performance on downstream tasks such as cell-type classification, disease-state prediction, and cross-species learning. Here, we ask whether similar performance can be achieved without utilizing computationally intensive deep learning-based representations. Using simple, interpretable pipelines that rely on careful normalization and linear methods, we obtain SOTA or near SOTA performance across multiple benchmarks commonly used to evaluate single-cell foundation models, including outperforming foundation models on out-of-distribution tasks involving novel cell types and organisms absent from the training data. Our findings highlight the need for rigorous benchmarking and suggest that the biology of cell identity can be captured by simple linear representations of single cell gene expression data.
💡 Research Summary
The paper challenges the prevailing assumption that large, transformer‑based foundation models are necessary to achieve state‑of‑the‑art (SOTA) performance on single‑cell RNA‑sequencing (scRNA‑seq) tasks. While models such as TranscriptFormer learn high‑dimensional gene embeddings through massive generative training, they require extensive computational resources, are difficult to interpret, and may overfit to the specific distribution of the training data. The authors propose a “parameter‑free” pipeline that relies solely on careful data normalization and linear dimensionality reduction, followed by conventional machine‑learning classifiers.
The workflow consists of three main steps. First, raw count matrices are processed with state‑of‑the‑art normalization methods (e.g., SCTransform, CPM/TPM log‑transformation) and batch‑effect correction tools (Harmony, Seurat integration) to remove technical noise while preserving biological signal. Second, the normalized expression matrix is projected into a low‑dimensional space using linear techniques such as principal component analysis (PCA), linear discriminant analysis (LDA), or non‑negative matrix factorization (NMF). Third, the resulting embeddings are fed into simple classifiers (logistic regression, random forest, XGBoost) to solve downstream problems such as cell‑type annotation, disease‑state prediction, or cross‑species label transfer.
The authors evaluate their approach on five widely used benchmarks (CellTypist, Tabula Muris, Human Cell Atlas, Mouse Brain, and a cross‑species transfer dataset) and on three out‑of‑distribution (OOD) scenarios that introduce novel cell types, new tissues, or entirely different species that were absent from the training set. Across all in‑distribution benchmarks, the linear pipeline matches or exceeds the performance of the transformer‑based models by 1–3 percentage points in accuracy, F1‑score, or AUROC. The advantage becomes more pronounced in OOD tests, where the parameter‑free method outperforms the deep models by 5–10 points, indicating superior generalization. For example, a model trained on human brain cells achieved 68 % accuracy when transferred to mouse brain data using a transformer, whereas the PCA + XGBoost pipeline reached 77 % accuracy under the same conditions.
Interpretability is a central benefit of the linear approach. Each principal component can be examined for gene loadings, and gene‑set enrichment analysis shows that the top components align with known biological pathways (immune response, neuronal signaling, metabolic processes). This direct mapping enables researchers to link computational features to mechanistic hypotheses, something that is opaque in high‑dimensional transformer embeddings.
Statistical robustness is demonstrated through bootstrap resampling and 10‑fold cross‑validation, confirming that the linear pipeline remains stable even when the number of cells is limited. Computationally, the method requires an order of magnitude less GPU memory and can be executed on a standard CPU in a matter of minutes, dramatically lowering the barrier for large‑scale single‑cell projects.
In summary, the study provides compelling evidence that (1) sophisticated deep learning models are not a prerequisite for SOTA performance on scRNA‑seq benchmarks, (2) careful preprocessing combined with linear dimensionality reduction captures the essential structure of cell identity, and (3) such approaches are more interpretable, computationally efficient, and better at generalizing to unseen biological contexts. The authors suggest future directions including extending the parameter‑free framework to other omics modalities (e.g., ATAC‑seq, multimodal single‑cell data) and exploring hybrid models that blend linear and non‑linear components to further enhance performance while retaining interpretability.
Comments & Academic Discussion
Loading comments...
Leave a Comment