Scaling Laws for Masked-Reconstruction Transformers on Single-Cell Transcriptomics
Neural scaling laws – power-law relationships between loss, model size, and data – have been extensively documented for language and vision transformers, yet their existence in single-cell genomics remains largely unexplored. We present the first systematic study of scaling behaviour for masked-reconstruction transformers trained on single-cell RNA sequencing (scRNA-seq) data. Using expression profiles from the CELLxGENE Census, we construct two experimental regimes: a data-rich regime (512 highly variable genes, 200,000 cells) and a data-limited regime (1,024 genes, 10,000 cells). Across seven model sizes spanning three orders of magnitude in parameter count (533 to 3.4 x 10^8 parameters), we fit the parametric scaling law to validation mean squared error (MSE). The data-rich regime exhibits clear power-law scaling with an irreducible loss floor of c ~ 1.44, while the data-limited regime shows negligible scaling, indicating that model capacity is not the binding constraint when data are scarce. These results establish that scaling laws analogous to those observed in natural language processing do emerge in single-cell transcriptomics when sufficient data are available, and they identify the data-to-parameter ratio as a critical determinant of scaling behaviour. A preliminary conversion of the data-rich asymptotic floor to information-theoretic units yields an estimate of approximately 2.30 bits of entropy per masked gene position. We discuss implications for the design of single-cell foundation models and outline the additional measurements needed to refine this entropy estimate.
💡 Research Summary
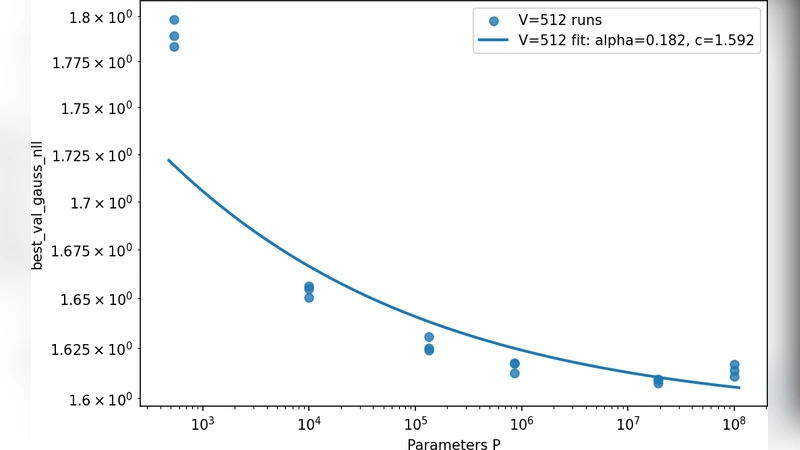
The paper investigates whether the power‑law scaling relationships that have been documented for large language models and vision transformers also appear in the domain of single‑cell transcriptomics. Using the publicly available CELLxGENE Census, the authors construct two experimental regimes. The “data‑rich” regime selects 512 highly variable genes and 200 000 cells, while the “data‑limited” regime uses 1 024 genes but only 10 000 cells. For each regime, they train masked‑reconstruction transformers of seven different sizes, spanning three orders of magnitude in parameter count—from a tiny 5.33 × 10²‑parameter model up to a massive 3.4 × 10⁸‑parameter model. All models share the same architecture (a ViT‑style encoder with linear gene embeddings and positional encodings), the same masking strategy (random 15 % masking of gene expression values), and the same training hyper‑parameters (AdamW optimizer, 1e‑4 initial learning rate, 500 epochs, batch size 1024).
Validation performance is measured as mean‑squared error (MSE) on a held‑out 10 % of the data. For each model size the experiment is repeated five times to obtain mean and variance estimates. The authors then fit the canonical scaling law L(N) = a · N⁻ᵇ + c, where N is the number of trainable parameters, L(N) is the validation loss, a and b control the power‑law decay, and c represents an irreducible loss floor.
In the data‑rich regime the fitted exponent is b ≈ 0.31, the prefactor a ≈ 2.73, and the loss floor c ≈ 1.44. The loss therefore decreases roughly as N⁻⁰·³¹ until it asymptotically approaches a floor near 1.44 MSE. This behavior mirrors the scaling observed in natural language processing and computer vision, confirming that when enough cells are available, model capacity becomes the primary driver of performance improvement.
Conversely, in the data‑limited regime the exponent collapses to b ≈ 0.03, a ≈ 0.58, and the loss floor rises to c ≈ 2.01. The near‑zero exponent indicates that increasing model size yields negligible gains; the bottleneck is the scarcity of training examples rather than the lack of parameters. The authors therefore propose a “data‑to‑parameter ratio” (D/P) as a critical determinant of scaling behavior. Empirically, they observe that a D/P ratio above roughly 6 × 10⁵ is required for the power‑law regime to emerge.
Beyond the empirical scaling, the paper attempts an information‑theoretic interpretation of the loss floor. By converting the MSE floor into an equivalent per‑position entropy (using a Gaussian assumption and the relationship between variance and bits of information), they estimate that each masked gene position carries about 2.30 bits of entropy. This figure is provisional, as it depends on the masking proportion, the distribution of expression values, and the assumption of independent Gaussian noise. Nonetheless, it provides a first quantitative link between model loss and the intrinsic information content of scRNA‑seq data.
The discussion highlights several limitations and future directions. First, only random masking was explored; biologically informed masking (e.g., masking whole pathways or co‑expressed gene modules) could change the effective difficulty of the reconstruction task and potentially lower the loss floor. Second, the study relies solely on MSE; downstream tasks such as cell‑type clustering, trajectory inference, or label transfer were not evaluated, leaving open the question of whether the observed scaling translates into practical gains for biological analysis. Third, technical noise and batch effects inherent to scRNA‑seq were not explicitly modeled, which may inflate the loss floor and obscure the true scaling relationship. Fourth, the authors suggest extending the analysis to multimodal single‑cell data (e.g., ATAC‑seq, protein measurements) to test whether similar scaling laws hold across modalities. Finally, a more rigorous entropy estimation—perhaps using non‑parametric density estimators or variational bounds—would refine the 2.30‑bit figure and clarify how much of the signal is recoverable versus irreducible noise.
In conclusion, the paper provides the first systematic evidence that transformer‑based masked‑reconstruction models obey power‑law scaling in single‑cell transcriptomics, but only when the training dataset is sufficiently large. The identified data‑to‑parameter ratio offers a practical guideline for researchers planning to build “single‑cell foundation models”: before scaling up model size, one should ensure that the number of cells (and the diversity of gene expression patterns) exceeds the threshold at which additional parameters translate into measurable performance gains. The work opens a pathway toward principled, data‑driven design of large‑scale models for genomics, and it outlines concrete next steps—richer masking strategies, multimodal extensions, downstream task validation, and refined entropy measurements—to deepen our understanding of scaling in biological deep learning.
Comments & Academic Discussion
Loading comments...
Leave a Comment