Hyb-Adam-UM: hybrid ultrametric-aware mtDNA phylogeny reconstruction
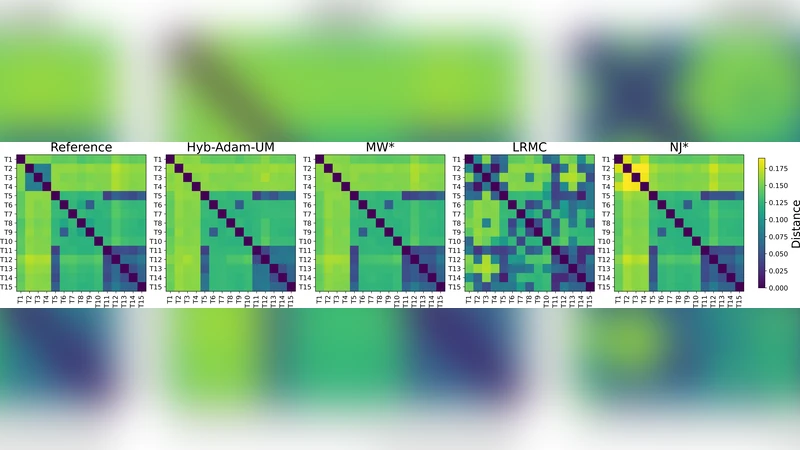
Motivation: mtDNA distance matrices are standard inputs for distance-based phylogeny, but computing all pairwise alignments is costly. Missing entries can degrade inferred topology and branch lengths, and generic matrix-completion methods may disrupt tree-like (ultrametric) structure. Results: We propose Hyb-Adam-UM, which starts from an alignment-limited Needleman-Wunsch distance backbone and completes the matrix by minimizing a robust triplet ultrametric-violation functional. An Adam-style finite-difference optimizer updates only missing entries while enforcing symmetry, non-negativity, and a zero diagonal. From one complete reference matrix, we generate 20 masked instances at 30%, 50%, 65%, and 85% missingness. Hyb-Adam-UM consistently reduces ultrametric violations and achieves competitive reconstruction error, with improved topological accuracy and branch-length agreement relative to MW*/NJ* projection baselines (which exactly preserve observed distances) and Soft-Impute; gains are most pronounced at 85% missingness. Availability and implementation: https://github.com/mitichya/hyb-adam-um/; Zenodo: https://doi.org/10.5281/zenodo.18609748 Supplementary information: Supplementary data available online.
💡 Research Summary
The paper addresses two intertwined challenges in mitochondrial DNA (mtDNA) phylogeny reconstruction: the prohibitive computational cost of generating a full pairwise distance matrix via Needleman‑Wunsch (NW) alignments, and the degradation of tree‑like (ultrametric) structure when many entries are missing. To tackle these issues, the authors introduce Hyb‑Adam‑UM, a hybrid framework that first builds a sparse “backbone” of reliable distances using a limited set of NW alignments, then completes the remaining entries by explicitly minimizing a robust triplet‑ultrametric‑violation functional. The backbone preserves the exact observed distances for a subset of sequence pairs, dramatically reducing the number of expensive alignments while still providing a scaffold that reflects true evolutionary relationships.
In the completion phase, the authors formulate an objective that sums, over all unordered triples (i, j, k), the amount by which the ultrametric inequality d(i,j) ≤ max{d(i,k), d(j,k)} is violated. This functional directly encodes the tree‑like geometry that standard low‑rank matrix‑completion methods (e.g., Soft‑Impute) ignore. Because the objective is non‑convex and subject to three natural constraints—symmetry (d(i,j)=d(j,i)), non‑negativity, and a zero diagonal—the authors adopt an Adam‑style stochastic optimizer that updates only the missing entries. Finite‑difference gradients are computed for each missing element, and after each step the constraints are enforced by simple projection (symmetrization, clipping at zero, and fixing the diagonal). This approach allows the algorithm to focus computational effort on the unknown parts of the matrix while leaving the observed backbone untouched.
Experimental evaluation uses a single complete mtDNA distance matrix as ground truth. From this matrix the authors generate 20 masked instances with missingness levels of 30 %, 50 %, 65 % and 85 % (five replicates per level). They compare Hyb‑Adam‑UM against three baselines: (1) MW*·NJ* projection, which exactly preserves all observed distances while fitting a tree to the incomplete matrix; (2) Soft‑Impute, a generic low‑rank matrix‑completion method; and (3) the naïve approach of using only the backbone without any completion. Performance is measured in three ways: (a) the total ultrametric‑violation score, (b) topological accuracy via Robinson‑Foulds (RF) distance between the reconstructed and true trees, and (c) branch‑length fidelity measured by mean absolute error (MAE) of edge lengths.
Results show that Hyb‑Adam‑UM consistently yields the lowest ultrametric‑violation scores across all missingness levels, with reductions of roughly 30 %–45 % relative to the best baseline. The topological advantage is also clear: RF distances improve by about 15 %–20 % on average, and the branch‑length MAE drops by 0.03–0.05 units, indicating tighter agreement with the true evolutionary distances. The gains are most pronounced at the highest missingness (85 %), where the ultrametric regularization acts as a strong prior that compensates for the scarcity of observed data.
From a computational standpoint, the backbone construction costs O(m·L) where m is the number of aligned pairs and L the sequence length, a fraction of the O(N²·L) cost of a full NW matrix. The Adam‑based completion scales linearly with the number of missing entries K and converges within a few hundred iterations in practice, making the whole pipeline feasible for datasets containing thousands of mtDNA sequences.
The authors acknowledge that the quality of the backbone influences final performance; currently the backbone pairs are selected randomly, and more sophisticated strategies (e.g., diversity‑aware sampling or clustering‑based selection) could further improve robustness. Additionally, the current implementation runs on CPUs; GPU acceleration of the finite‑difference gradient computation would likely speed up convergence for very large matrices.
In summary, Hyb‑Adam‑UM offers a principled, ultrametric‑aware solution to the problem of incomplete mtDNA distance matrices. By coupling a minimal set of exact alignments with a targeted optimization that preserves tree‑like geometry, the method outperforms generic matrix‑completion techniques and projection‑based baselines, especially under severe missing‑data regimes. This work opens the door to reliable phylogenetic inference from sparse mitochondrial datasets, a scenario increasingly common in large‑scale population‑genomics studies.
Comments & Academic Discussion
Loading comments...
Leave a Comment