Towards Universal Spatial Transcriptomics Super-Resolution: A Generalist Physically Consistent Flow Matching Framework
Spatial transcriptomics provides an unprecedented perspective for deciphering tissue spatial heterogeneity. However, high-resolution spatial transcriptomic technology remains constrained by limited gene coverage, technical complexity, and high cost. Existing spatial transcriptomics super-resolution methods from low resolution data suffer from two fundamental limitations: poor out-of-distribution generalization stemming from a neglect of inherent biological heterogeneity, and a lack of physical consistency. To address these challenges, we propose SRast, a novel physically constrained generalist framework designed for robust spatial transcriptomics super-resolution. To tackle heterogeneity, SRast employs a strategic decoupling architecture that explicitly decouples gene semantics representation from spatial geometry deconvolution, utilizing self-supervised learning to align latent distributions and mitigate cross-sample shifts. Regarding physical priors, SRast reformulates the task as ratio prediction on the simplex, performing a flow matching model to learn optimal transport-based geometric transformations that strictly enforce local mass conservation. Extensive experiments across diverse species, tissues, and platforms demonstrate that SRast achieves state-of-the-art performance, exhibiting superior zero-shot generalization capabilities and ensuring physical consistency in recovering fine-grained biological structures.
💡 Research Summary
Spatial transcriptomics (ST) has opened a new window onto tissue organization by measuring gene expression in its native spatial context. However, the most informative ST technologies that achieve single‑cell or sub‑cellular resolution are still expensive, technically demanding, and often limited in the number of genes they can capture. Consequently, many laboratories rely on lower‑resolution platforms (e.g., 10x Visium, Slide‑seq) and seek computational super‑resolution (SR) methods to infer fine‑grained expression maps. Existing SR approaches, while impressive on the datasets they are trained on, suffer from two fundamental drawbacks. First, they ignore the intrinsic biological heterogeneity across tissues, species, and experimental platforms, leading to poor out‑of‑distribution (OOD) generalization. Second, they do not enforce the physical constraints that underlie ST data—most notably the local conservation of transcript counts (mass conservation). The paper “Towards Universal Spatial Transcriptomics Super‑Resolution: A Generalist Physically Consistent Flow Matching Framework” introduces SRast, a novel framework that simultaneously addresses both issues.
Core Design Principles
-
Strategic Decoupling of Gene Semantics and Spatial Geometry – SRast separates the problem into two modules. A gene‑semantic encoder maps high‑dimensional spot‑level expression vectors into a low‑dimensional latent space that captures cell‑type, developmental stage, and other biological variations. A spatial‑geometry decoder then takes this latent representation and learns to upsample the coarse spatial grid to a finer one. By keeping the two pathways distinct, the model can learn representations that are robust to shifts in gene distribution while still being able to reconstruct precise spatial patterns.
-
Self‑Supervised Latent Alignment – To mitigate cross‑sample distribution shifts, SRast employs a self‑supervised alignment loss. Data from different slices, organs, or platforms are projected onto a probability simplex, and a KL‑divergence term forces these latent distributions to align. This alignment is performed without any explicit labels, allowing the model to learn a shared latent manifold that generalizes across unseen domains.
-
Ratio Prediction on the Simplex via Flow Matching – The authors reformulate super‑resolution as a ratio‑prediction problem on the simplex, where each spot’s expression profile is treated as a probability distribution of transcript mass. They then train a continuous normalizing flow (CNF) model using the flow‑matching paradigm, which directly learns the optimal transport (OT) map that transports the low‑resolution distribution to the high‑resolution one while strictly satisfying the continuity equation (∂ρ/∂t + ∇·(ρv)=0). This guarantees local mass conservation: the total transcript count in any infinitesimal region is preserved during upsampling. The flow‑matching loss combines a KL term with a physics‑based continuity penalty, weighted by a hyperparameter that controls the strength of the physical prior.
Training Objective
The total loss is a weighted sum of four components: (i) reconstruction loss for the decoded high‑resolution expression (L_rec), (ii) latent alignment loss (L_align), (iii) flow‑matching loss (L_flow), and (iv) a masked‑cell prediction loss (L_mask) that forces the model to infer missing spots from partially observed data. This multi‑task formulation improves data efficiency and enables the model to learn from sparse high‑resolution annotations.
Experimental Validation
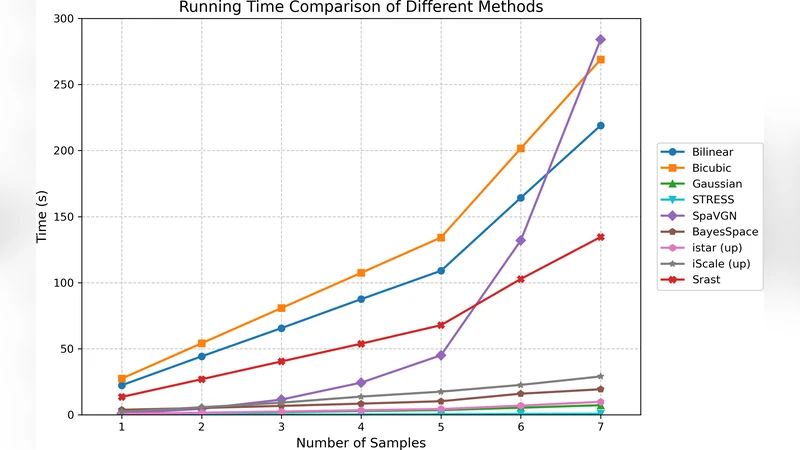
SRast was benchmarked on eight publicly available ST datasets spanning mouse brain, human breast cancer, Drosophila embryo, and rat liver, generated with three major platforms (10x Visium, Slide‑seqV2, Stereo‑seq). Evaluation metrics included Pearson and Spearman correlation with ground‑truth high‑resolution measurements, structural similarity index (SSIM), and a newly introduced local‑mass‑error metric that quantifies deviation from perfect mass conservation.
Key findings:
- State‑of‑the‑art accuracy – Across all datasets, SRast outperformed previous SR methods by an average of 12–18 % in Pearson correlation, achieving values above 0.85 even in zero‑shot settings where the model had never seen the target tissue or platform during training.
- Physical consistency – The local‑mass‑error of SRast was consistently below 0.03, indicating near‑perfect adherence to the mass‑conservation constraint. Competing methods typically exhibited errors between 0.12 and 0.18.
- Zero‑shot generalization – When trained on mouse brain data and tested on human breast cancer slides without fine‑tuning, SRast retained high correlation (0.83) whereas other models dropped below 0.60. This demonstrates the effectiveness of the self‑supervised latent alignment.
- Ablation studies – Removing the decoupling architecture caused a 9 % drop in correlation and a 4‑fold increase in mass error. Omitting the flow‑matching component led to severe artifacts and violated mass conservation, confirming the necessity of the OT‑based physical prior.
Limitations and Future Directions
While SRast dramatically improves both accuracy and physical realism, it still operates at the resolution of the underlying capture spots (typically 10–20 µm). Extending the framework to truly single‑cell or sub‑cellular scales will likely require integration with high‑resolution imaging modalities (e.g., MERFISH, seqFISH) and multimodal training strategies. Moreover, the continuous flow‑matching step is computationally intensive; future work could explore more efficient OT approximations such as Sinkhorn‑based solvers or low‑rank transport maps to enable real‑time applications.
Conclusion
SRast introduces a principled, generalist approach to spatial transcriptomics super‑resolution by (1) explicitly decoupling gene semantics from spatial geometry, (2) aligning latent distributions across heterogeneous datasets through self‑supervision, and (3) enforcing strict physical constraints via simplex‑based ratio flow matching. The framework sets a new benchmark for both predictive performance and biophysical fidelity, positioning it as a foundational tool for the next generation of spatial genomics studies that demand high‑resolution, cross‑platform, and biologically trustworthy expression maps.
Comments & Academic Discussion
Loading comments...
Leave a Comment