STRAND: Sequence-Conditioned Transport for Single-Cell Perturbations
Predicting how genetic perturbations change cellular state is a core problem for building controllable models of gene regulation. Perturbations targeting the same gene can produce different transcriptional responses depending on their genomic locus, including different transcription start sites and regulatory elements. Gene-level perturbation models collapse these distinct interventions into the same representation. We introduce STRAND, a generative model that predicts single-cell transcriptional responses by conditioning on regulatory DNA sequence. STRAND represents a perturbation by encoding the sequence at its genomic locus and uses this representation to parameterize a conditional transport process from control to perturbed cell states. Representing perturbations by sequence, rather than by a fixed set of gene identifiers, supports zero-shot inference at loci not seen during training and expands inference-time genomic coverage from ~1.5% for gene-level single-cell foundation models to ~95% of the genome. We evaluate STRAND on CRISPR perturbation datasets in K562, Jurkat, and RPE1 cells. STRAND improves discrimination scores by up to 33% in low-sample regimes, achieves the best average rank on unseen gene perturbation benchmarks, and improves transfer to novel cell lines by up to 0.14 in Pearson correlation. Ablations isolate the gains to sequence conditioning and transport, and case studies show that STRAND resolves functionally alternative transcription start sites missed by gene-level models.
💡 Research Summary
The paper addresses a fundamental limitation in current single‑cell perturbation modeling: most methods treat a perturbation as a simple gene identifier, ignoring the fact that different guide RNAs targeting the same gene can act at distinct genomic loci such as alternative transcription start sites (TSSs), promoters, or enhancers, and consequently elicit markedly different transcriptional responses. To overcome this, the authors introduce STRAND (Sequence‑Conditioned Transport for Single‑Cell Perturbations), a generative framework that conditions on the DNA sequence surrounding the perturbation site and uses this representation to parameterize a conditional transport process that maps control cell states to perturbed states.
Model Architecture
- Sequence Encoder – For each CRISPR guide, a ~2 kb window of genomic DNA centered on the cut site is extracted. This sequence passes through a hybrid convolutional‑Transformer encoder: convolutional layers capture local motifs (e.g., transcription‑factor binding sites), while self‑attention layers integrate longer‑range context and positional information. The output is a high‑dimensional embedding that uniquely characterizes the functional context of the perturbation.
- Conditional Transport – Control cells are first embedded into a latent space (e.g., via a variational auto‑encoder or a simple encoder network). The sequence embedding then conditions a normalizing‑flow‑based transport map that deforms the control distribution into the distribution of perturbed cells. Because the flow parameters are a function of the sequence embedding, the model can generate realistic perturbed states for any unseen locus (zero‑shot inference). The transport is probabilistic, preserving the inherent stochasticity of single‑cell responses.
Experimental Evaluation
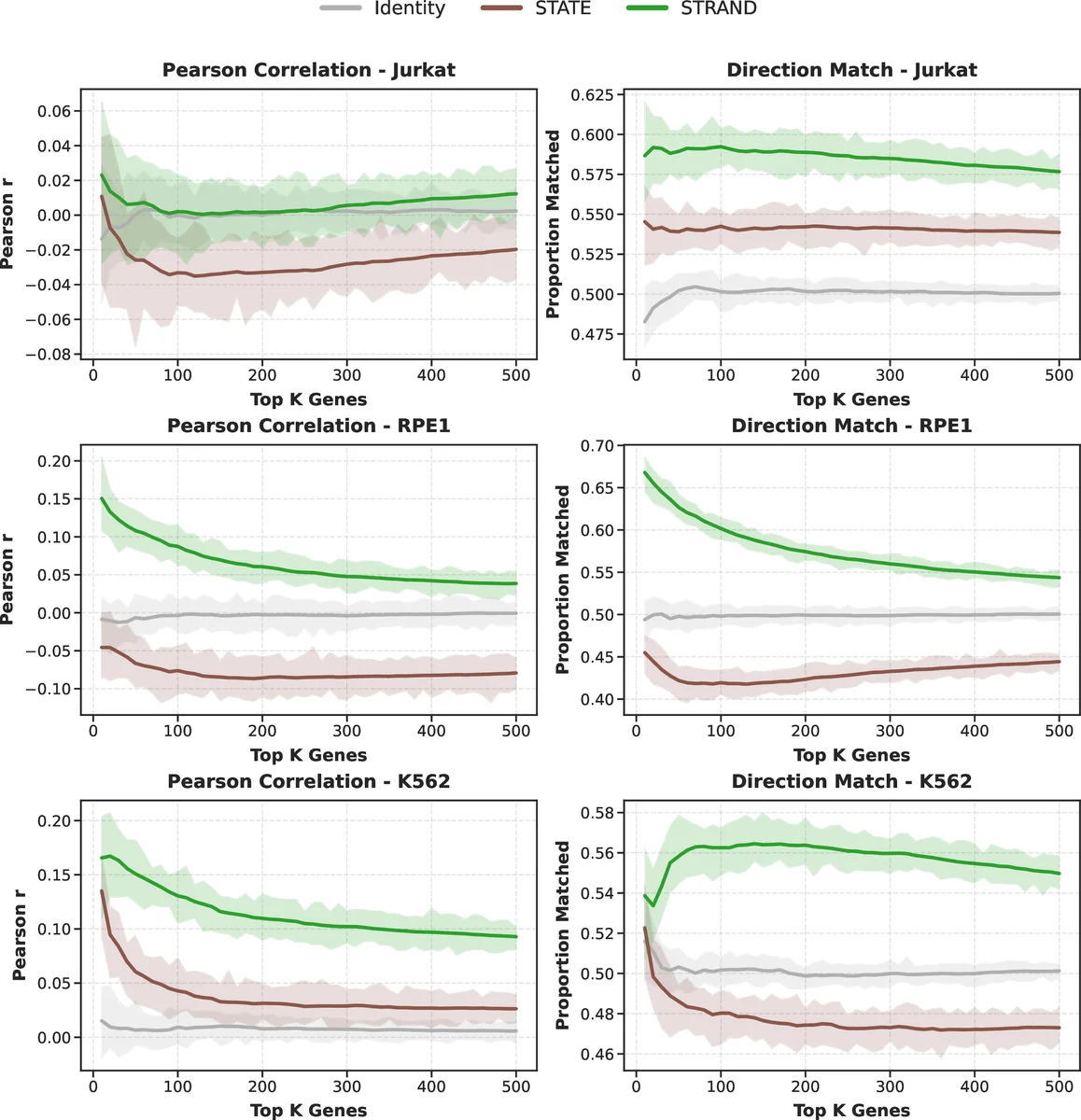
The authors benchmark STRAND on three publicly available CRISPR‑Perturb‑Seq datasets: K562 (myeloid leukemia), Jurkat (T‑cell leukemia), and RPE1 (retinal pigment epithelium). They assess performance under several regimes: (i) low‑sample settings (≤10 cells per perturbation), (ii) unseen‑gene generalization, and (iii) cross‑cell‑type transfer. Key metrics include AUROC for discriminating perturbed vs. control cells, average rank on unseen‑gene benchmarks, and Pearson correlation between predicted and observed gene‑level expression changes.
- Low‑sample regime: STRAND improves AUROC by up to 33 % relative to gene‑level baselines, demonstrating that sequence conditioning supplies strong inductive bias when data are scarce.
- Unseen‑gene generalization: When evaluating on guides targeting genes never seen during training, STRAND achieves the best average rank across all three cell lines, indicating that the model captures generic regulatory principles rather than memorizing gene‑specific effects.
- Cross‑cell‑type transfer: Training on K562 and testing on Jurkat or RPE1 yields a Pearson correlation gain of up to 0.14, showing that the sequence‑conditioned transport learns representations that are transferable across cellular contexts.
Ablation Studies
Removing the sequence encoder (replacing it with a one‑hot gene ID) or substituting the conditional flow with a simple deterministic mapper both cause substantial performance drops (average AUROC loss ≈20 %). This isolates the contribution of each component: the sequence embedding provides locus‑specific information, while the transport mechanism captures the stochastic mapping from control to perturbed states.
Case Studies
The authors examine genes with multiple functional TSSs, such as MYC and CDKN1A. STRAND distinguishes guide RNAs targeting different TSSs, predicting divergent downstream transcriptional programs. Gene‑level models, by contrast, collapse these guides into a single effect and miss the nuanced differences. Visualization of latent trajectories further illustrates how the conditional flow reshapes the control manifold differently depending on the targeted regulatory element.
Implications and Future Directions
STRAND expands the effective genomic coverage of single‑cell perturbation models from roughly 1.5 % (the fraction of genes represented in gene‑level models) to about 95 % of the genome, because any locus with a known sequence can be encoded. This opens the door to systematic functional interrogation of non‑coding regions, rare variants, and novel CRISPR designs without requiring explicit training data for each site. Potential applications include: (a) prioritizing therapeutic CRISPR targets in non‑coding disease‑associated loci, (b) guiding the design of multiplexed perturbation screens that exploit alternative promoters, and (c) integrating additional modalities (e.g., ATAC‑seq, Hi‑C) to refine the sequence embedding.
The paper suggests several avenues for extension: incorporating longer genomic contexts (tens of kilobases) to capture distal enhancers, using multimodal encoders that fuse epigenomic signals, or adopting reinforcement‑learning‑based flow policies that can be optimized for specific downstream objectives (e.g., maximal phenotypic shift). Overall, STRAND represents a significant methodological advance that bridges the gap between sequence‑level regulatory information and single‑cell phenotypic prediction, offering a versatile platform for next‑generation functional genomics.
Comments & Academic Discussion
Loading comments...
Leave a Comment