Smooth Dynamic Cutoffs for Machine Learning Interatomic Potentials
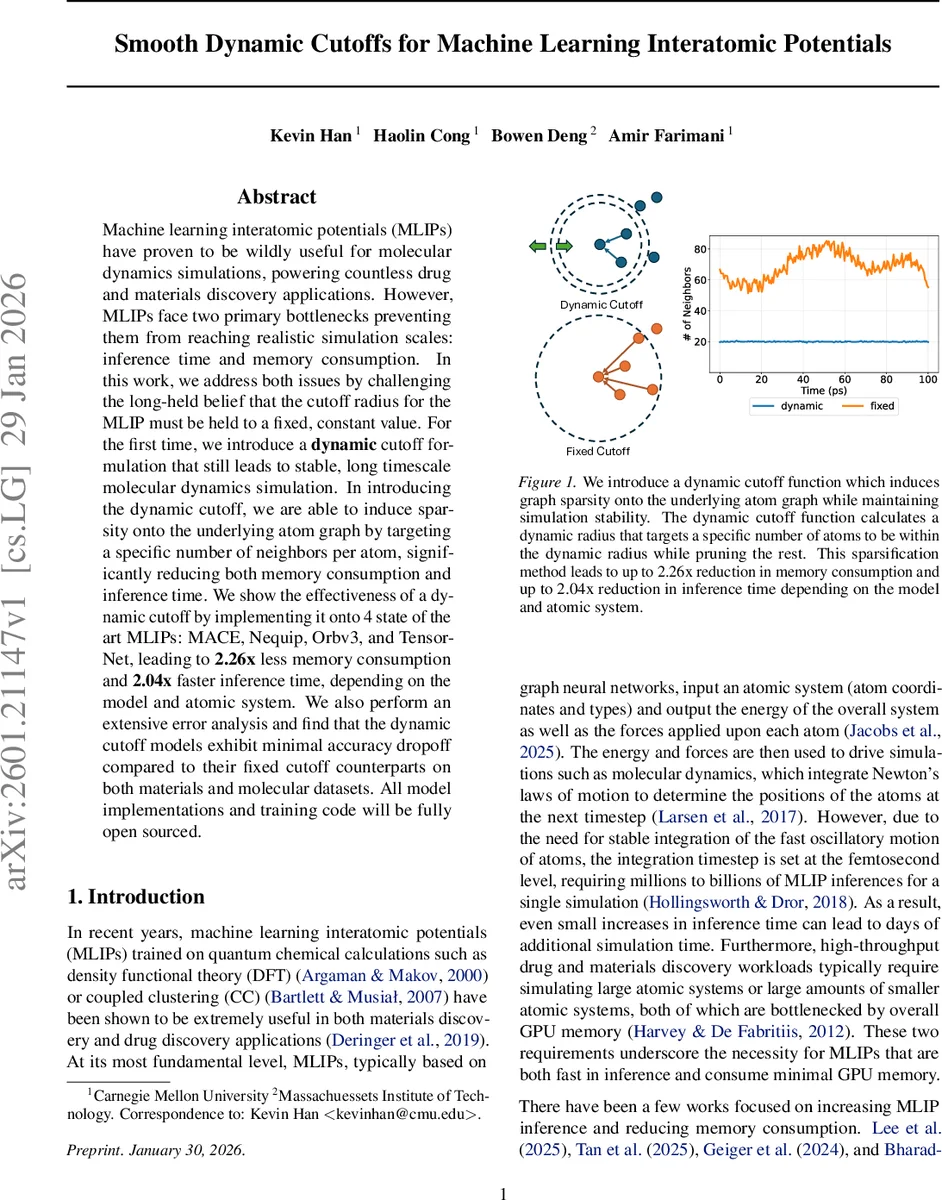
Machine learning interatomic potentials (MLIPs) have proven to be wildly useful for molecular dynamics simulations, powering countless drug and materials discovery applications. However, MLIPs face two primary bottlenecks preventing them from reaching realistic simulation scales: inference time and memory consumption. In this work, we address both issues by challenging the long-held belief that the cutoff radius for the MLIP must be held to a fixed, constant value. For the first time, we introduce a dynamic cutoff formulation that still leads to stable, long timescale molecular dynamics simulation. In introducing the dynamic cutoff, we are able to induce sparsity onto the underlying atom graph by targeting a specific number of neighbors per atom, significantly reducing both memory consumption and inference time. We show the effectiveness of a dynamic cutoff by implementing it onto 4 state of the art MLIPs: MACE, Nequip, Orbv3, and TensorNet, leading to 2.26x less memory consumption and 2.04x faster inference time, depending on the model and atomic system. We also perform an extensive error analysis and find that the dynamic cutoff models exhibit minimal accuracy dropoff compared to their fixed cutoff counterparts on both materials and molecular datasets. All model implementations and training code will be fully open sourced.
💡 Research Summary
Machine‑learning interatomic potentials (MLIPs) have become indispensable tools for accelerating molecular dynamics (MD) simulations in drug discovery, materials design, and many other domains. Despite their success, two fundamental bottlenecks limit their scalability: the time required for each inference step and the GPU memory needed to store the atom‑wise interaction graph. Existing work has tackled these issues by hand‑crafting CUDA kernels, pruning entire network layers, or distributing inference across multiple GPUs. However, none of these approaches fundamentally reduces the number of pairwise interactions that the model must process.
In this paper the authors challenge the long‑standing assumption that the cutoff radius in an MLIP must be a fixed, global constant. They introduce a dynamic cutoff that adapts locally for each atom, targeting a predefined average number of neighbors (μ) while preserving smoothness of the potential energy surface (PES). The method works in two stages. First, a conventional hard cutoff h is used to collect all neighbors of an atom v, forming the set N_v. Second, each neighbor u∈N_v is assigned a soft rank R_u based on its distance relative to all other neighbors. The rank is computed as a sum of sigmoid‑based indicators multiplied by a polynomial envelope p(x) that guarantees zero value and zero first‑ and second‑derivatives at the hard‑cutoff boundary. A Gaussian weighting function ω(R_u) = exp
Comments & Academic Discussion
Loading comments...
Leave a Comment