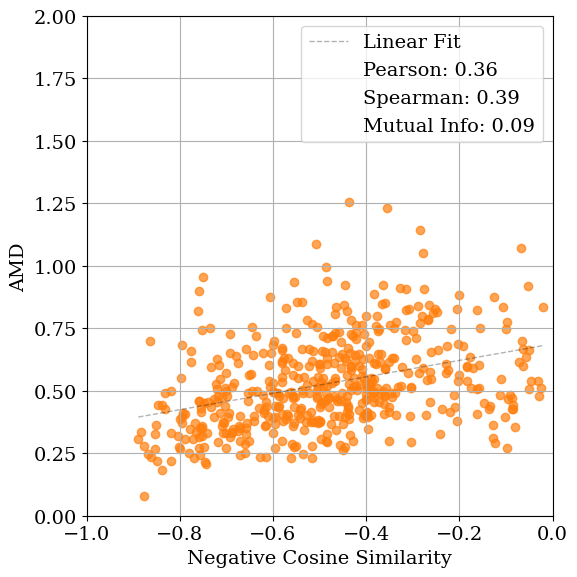
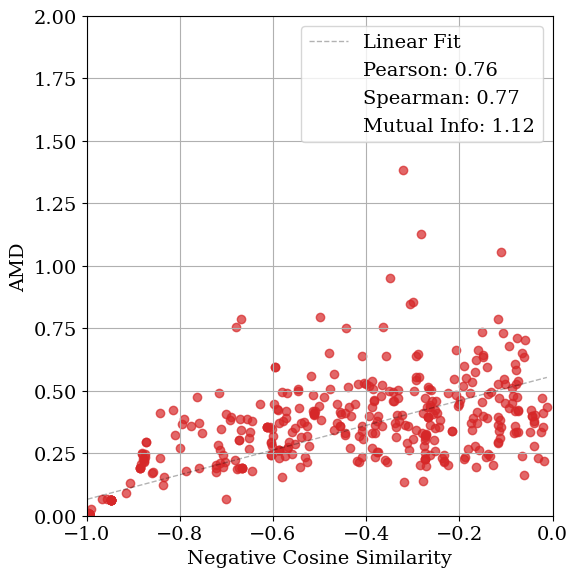
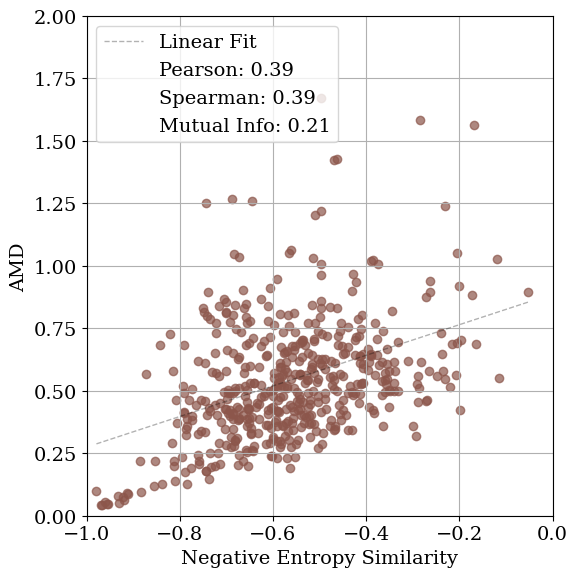
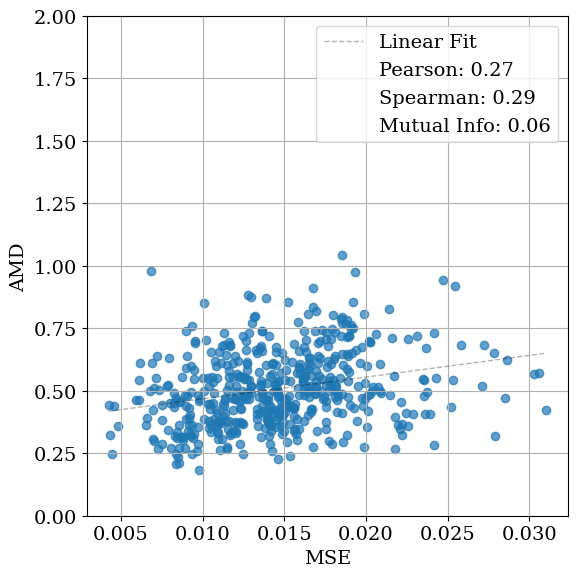
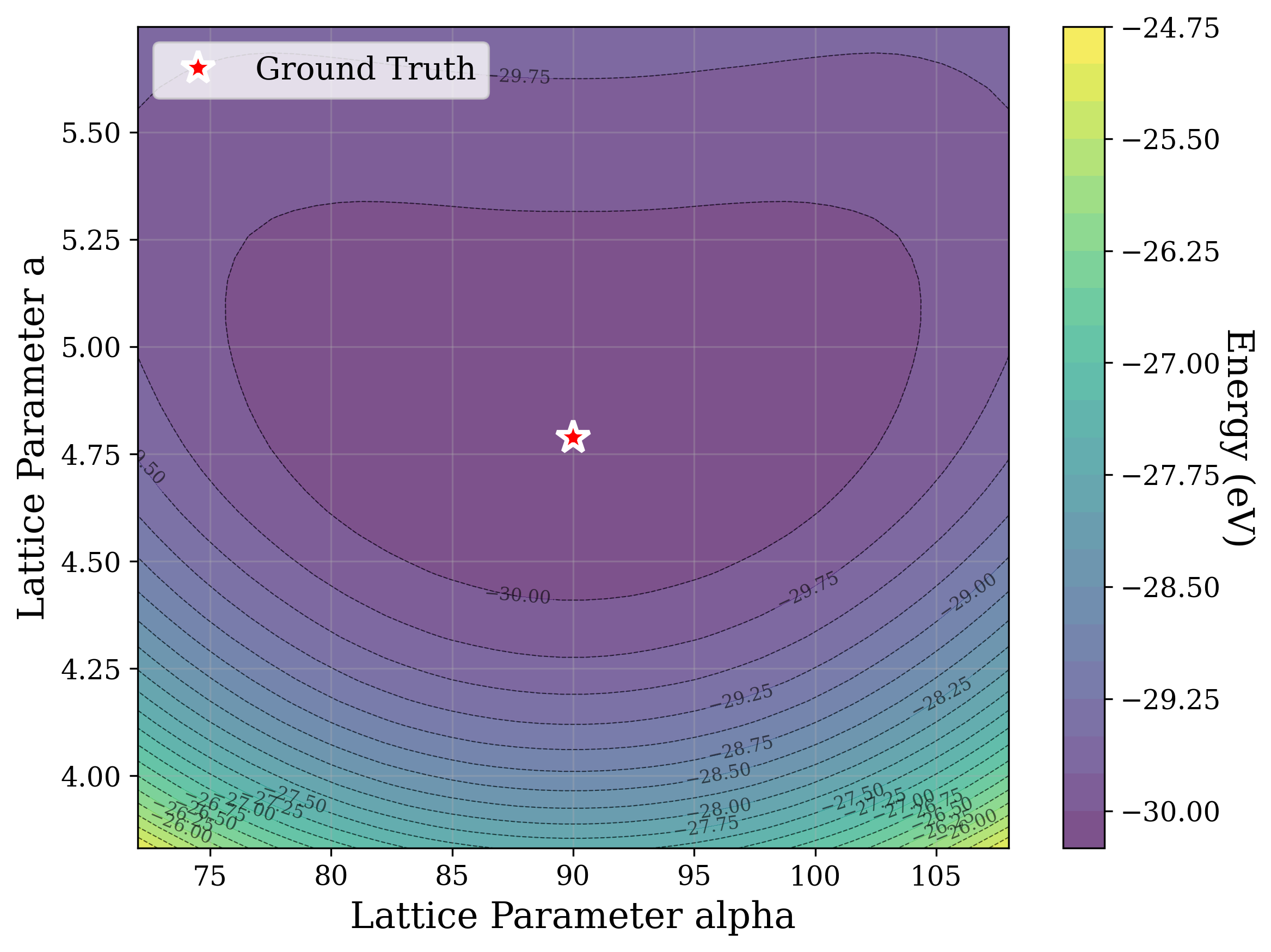
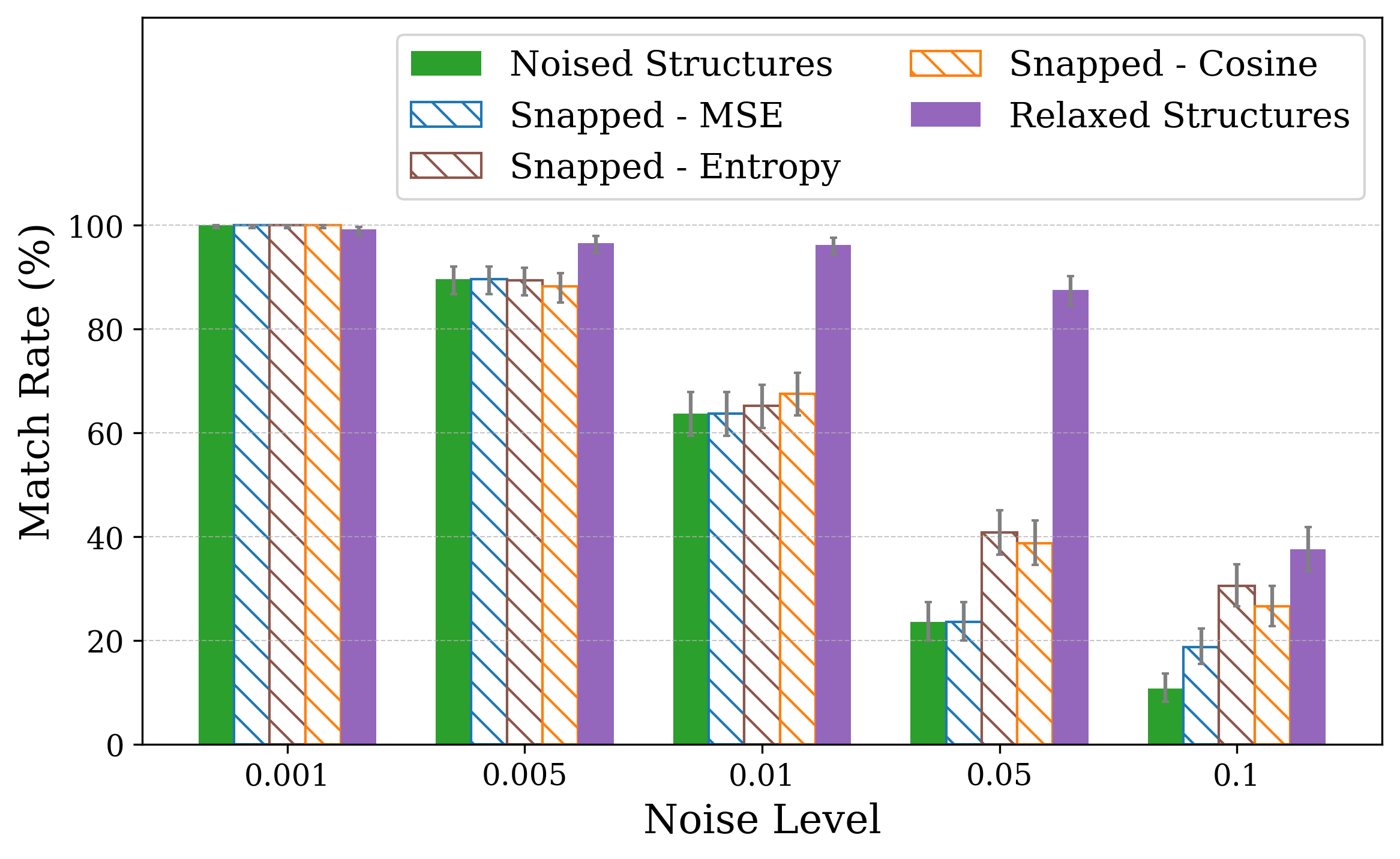
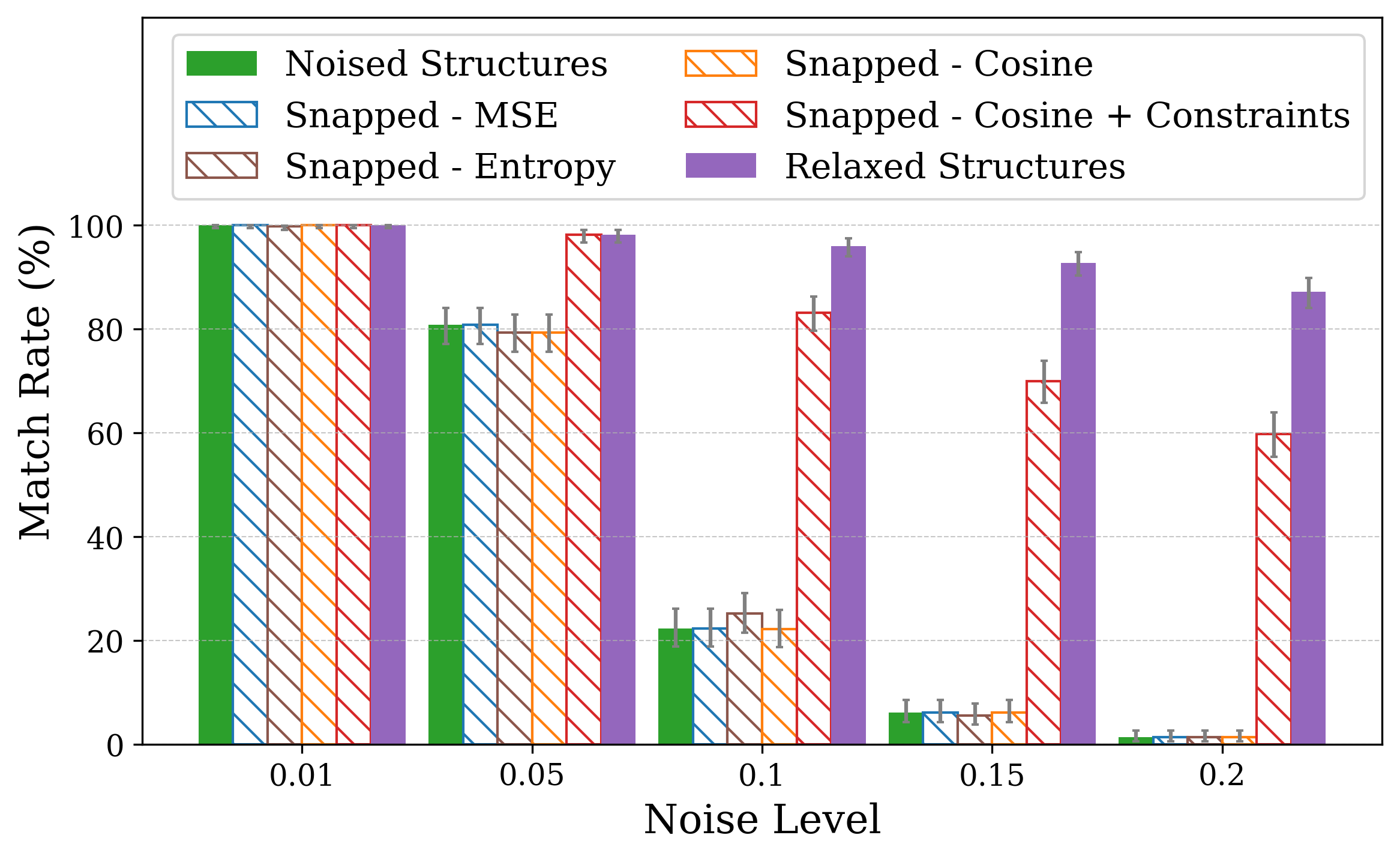
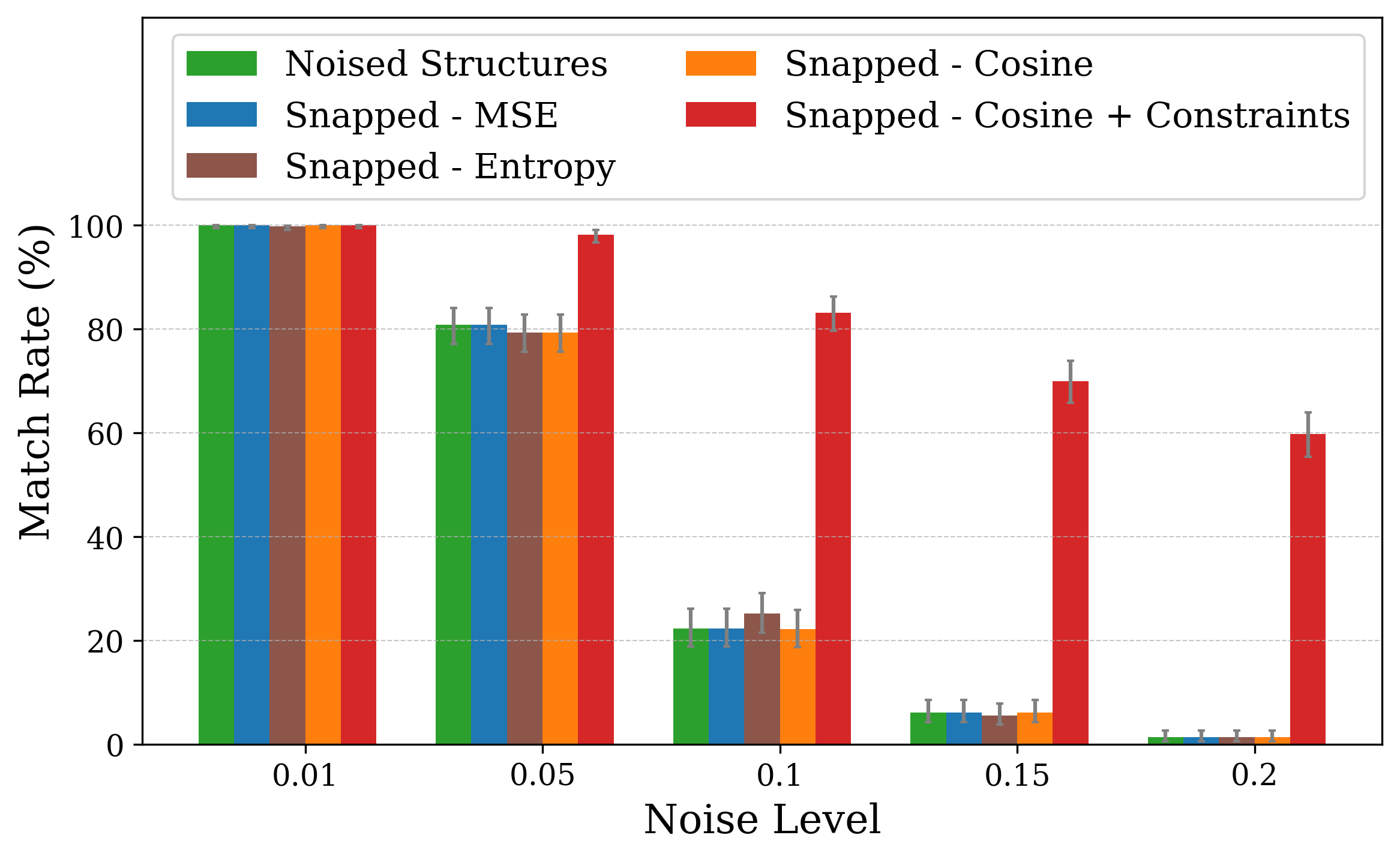
Solving crystal structures from powder X-ray diffraction (XRD) is a central challenge in materials characterization. In this work, we study the powder XRDto-structure mapping using gradient descent optimization, with the goal of recovering the correct structure from moderately distorted initial states based solely on XRD similarity. We show that commonly used XRD similarity metrics result in a highly non-convex landscape, complicating direct optimization. Constraining the optimization to the ground-truth crystal family significantly improves recovery, yielding higher match rates and increased mutual information and correlation scores between structural similarity and XRD similarity. Nevertheless, the landscape may remain non-convex along certain symmetry axes. These findings suggest that symmetry-aware inductive biases could play a meaningful role in helping learning models navigate the inverse mapping from diffraction to structure.
Deep Dive into The Loss Landscape of Powder X-Ray Diffraction-Based Structure Optimization Is Too Rough for Gradient Descent.
Solving crystal structures from powder X-ray diffraction (XRD) is a central challenge in materials characterization. In this work, we study the powder XRDto-structure mapping using gradient descent optimization, with the goal of recovering the correct structure from moderately distorted initial states based solely on XRD similarity. We show that commonly used XRD similarity metrics result in a highly non-convex landscape, complicating direct optimization. Constraining the optimization to the ground-truth crystal family significantly improves recovery, yielding higher match rates and increased mutual information and correlation scores between structural similarity and XRD similarity. Nevertheless, the landscape may remain non-convex along certain symmetry axes. These findings suggest that symmetry-aware inductive biases could play a meaningful role in helping learning models navigate the inverse mapping from diffraction to structure.
Determining the atomic structure of a crystal from its powder X-ray diffraction (XRD) pattern is a longstanding and central challenge in materials characterization [1,2]. The inverse problem of recovering the full three-dimensional crystal structure solely from an XRD pattern is extremely challenging due to the loss of phase information of the scattered waves -known as the phase problem [3] [4,Chapter 9.3,13.2]. Nevertheless, powder diffraction is widely used for identifying and characterizing crystalline solids. In practice, this is typically achieved by comparing the observed XRD spectrum to a reference database and performing least-squares refinement, known as Rietveld analysis [5][6][7]. However, this process is highly sensitive to initial parameters [8], and more importantly, it relies on the presence of the correct structure in the database and cannot be used to reconstruct novel or unreported phases.
Experimental phenomena such as preferred orientation, peak overlap, crystal twinning, and instrumental noise further complicate structural determination from powder XRD patterns [9][10][11]. Moreover, many minerals and metallic alloys exhibit solid solution ranges with very slight lattice shifts, which result in ranges of stoichiometries with nearly the same diffraction pattern [4,Chapter 10.3.2][ [12][13][14], making XRD-to-structure mapping a one-to-many problem. Consequently, accurate structure reconstruction typically requires refinement model fitting and domain-specific prior knowledge.
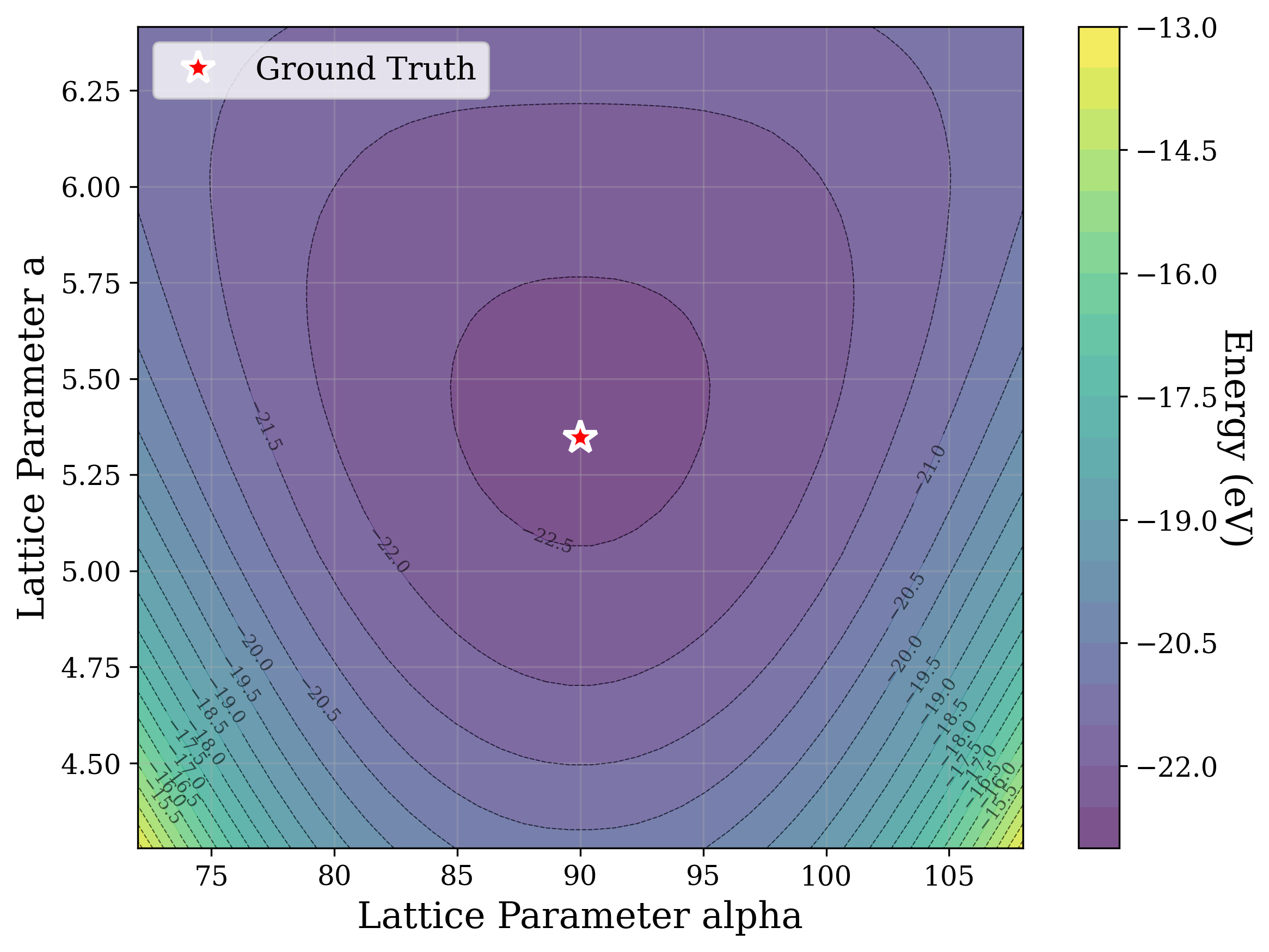
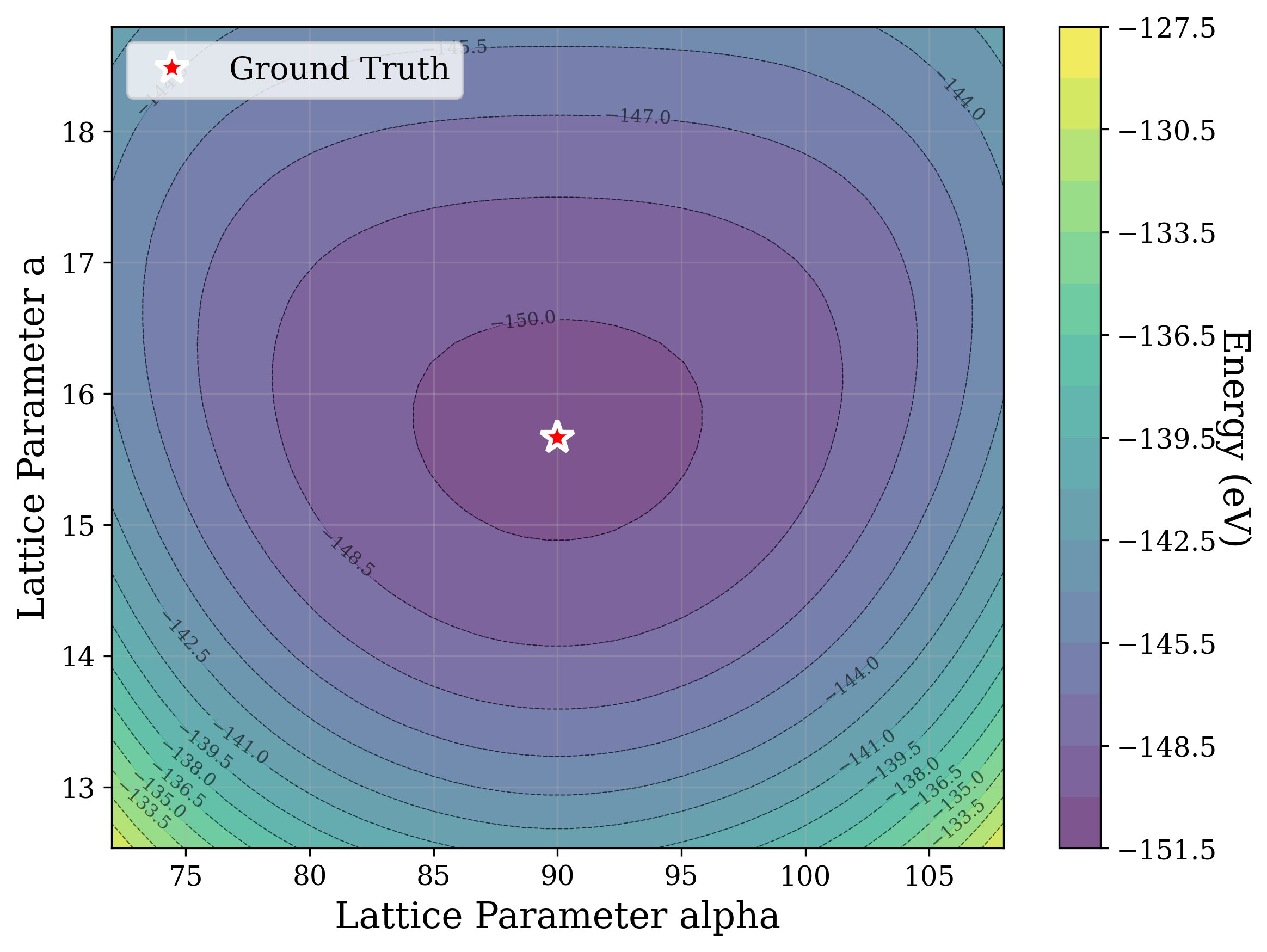
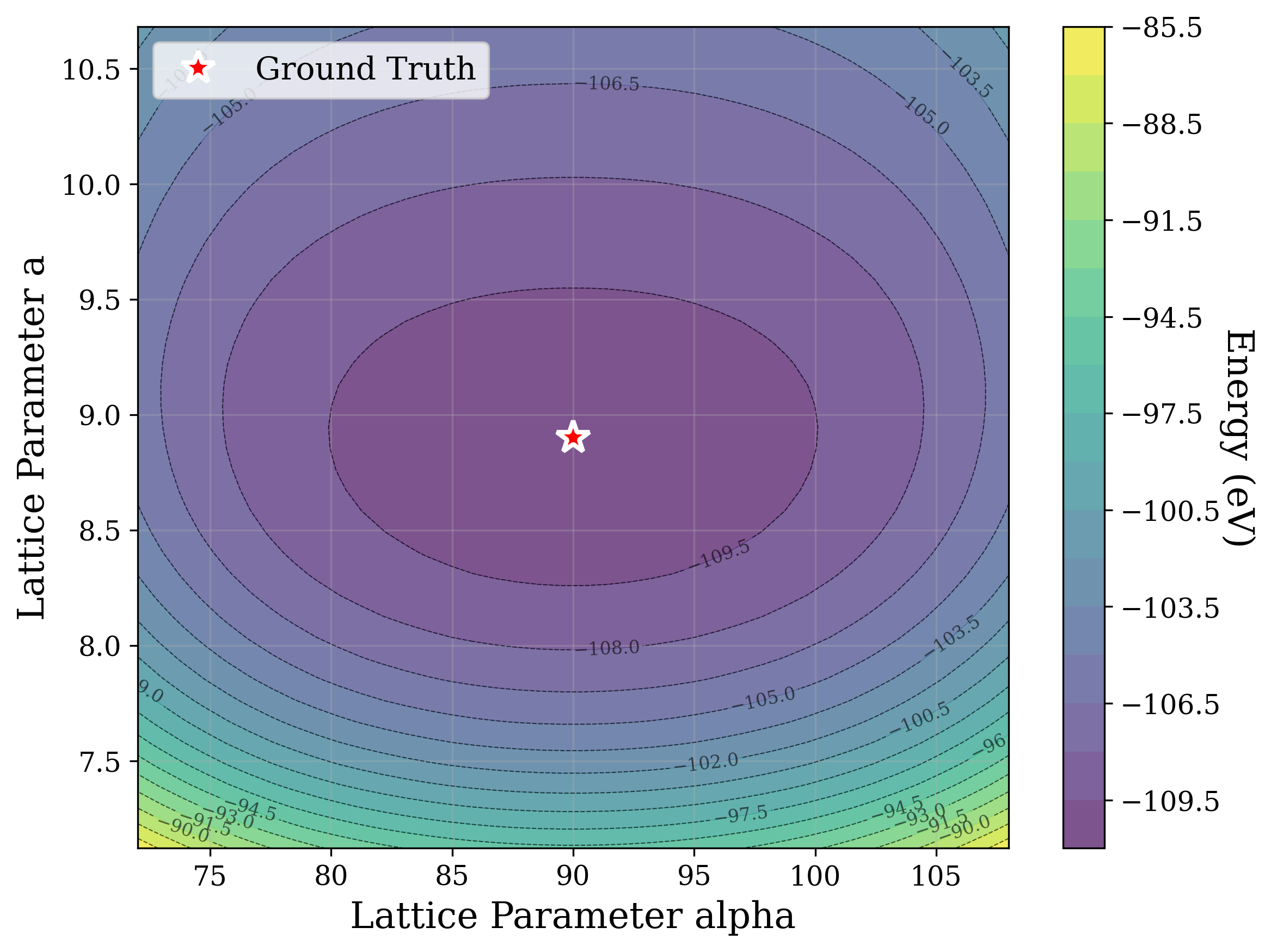
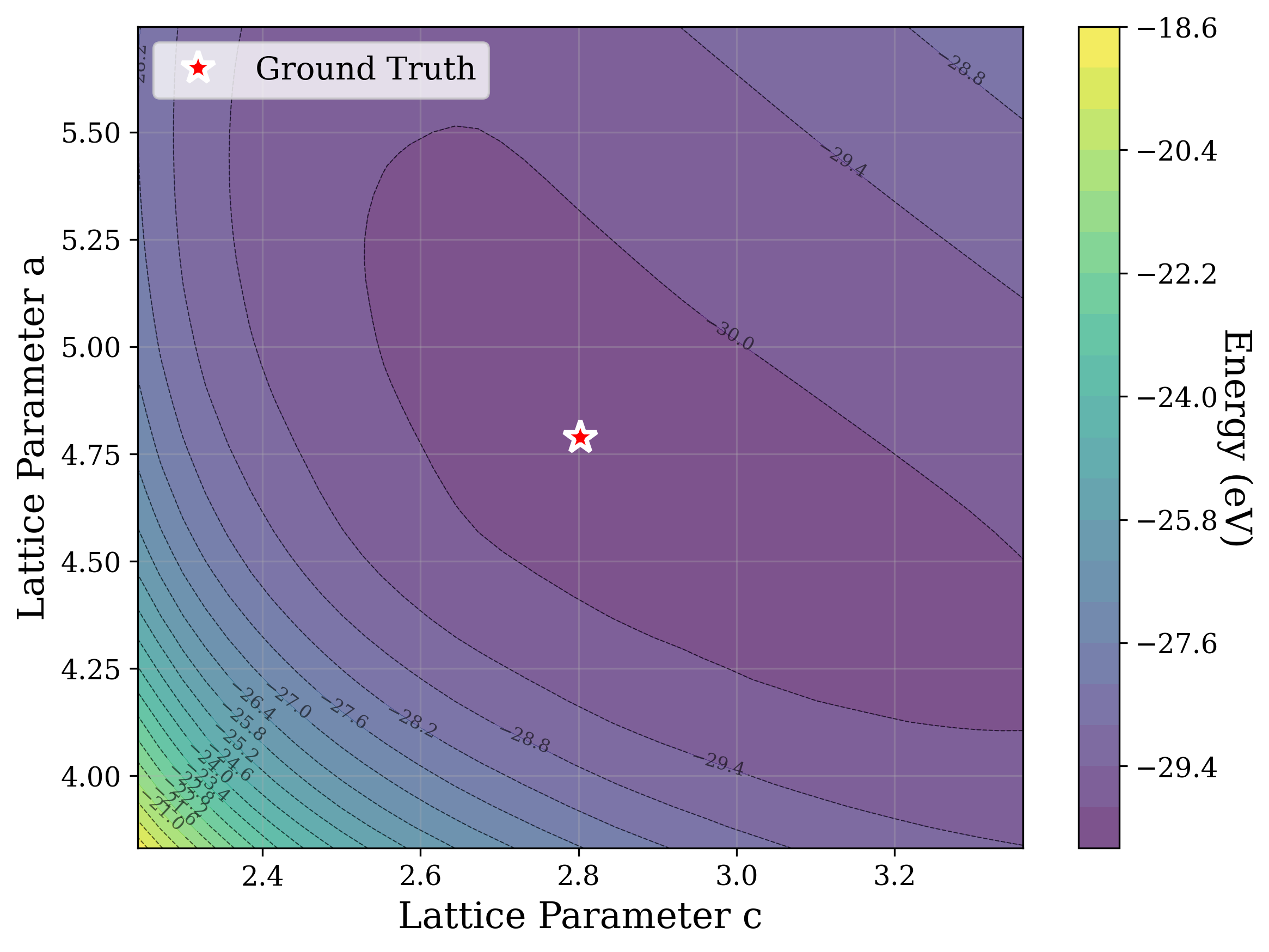
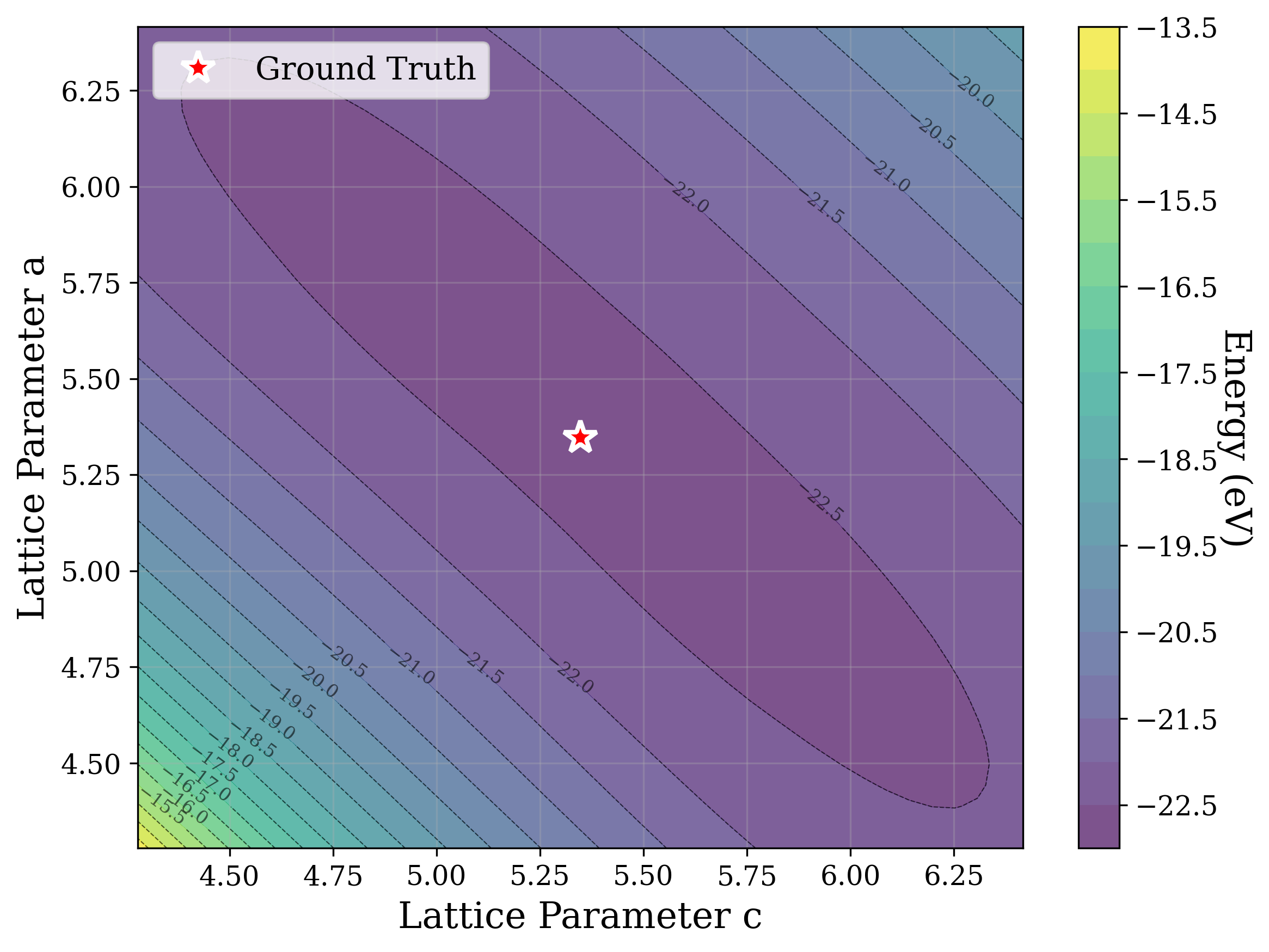
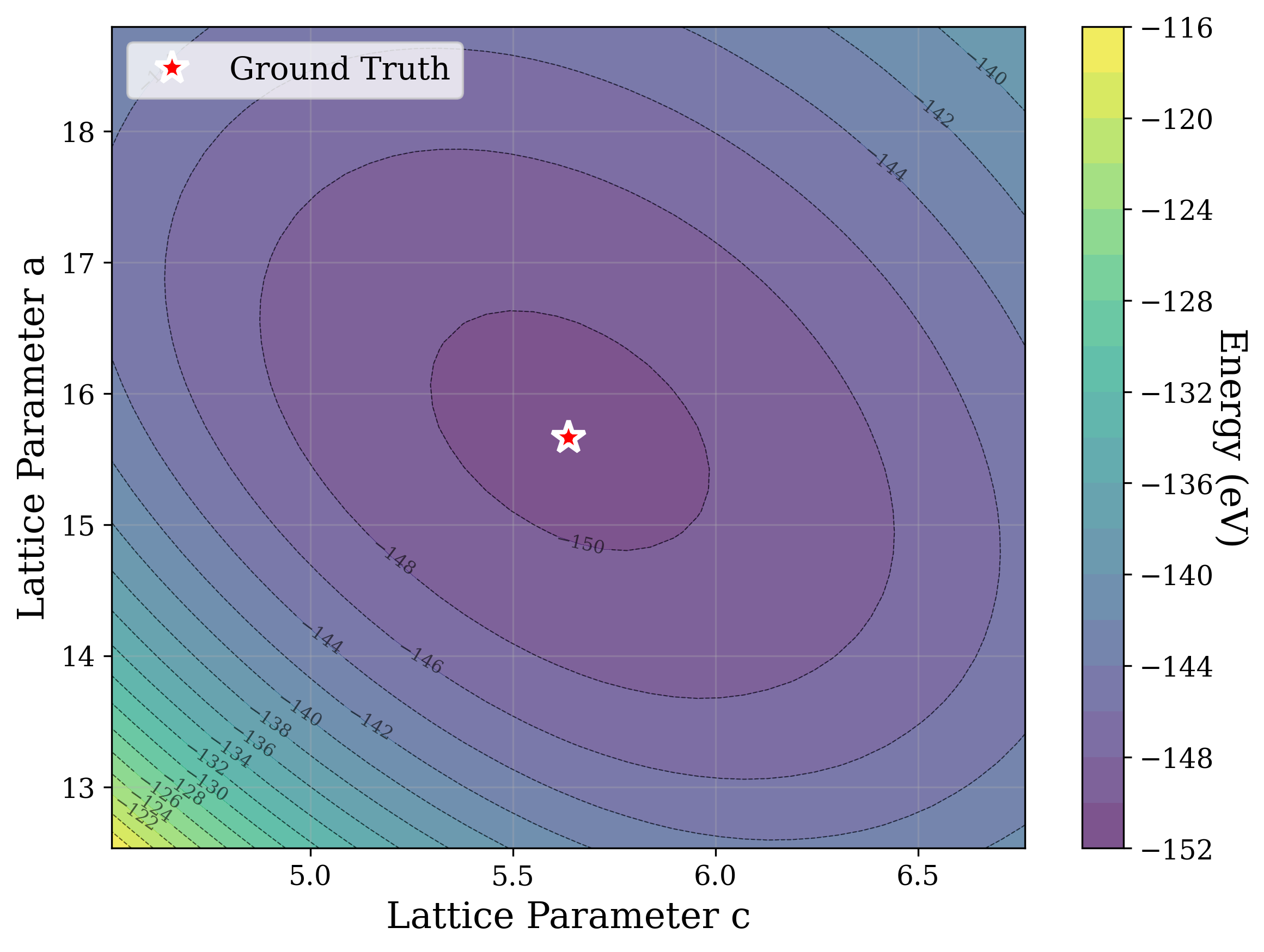
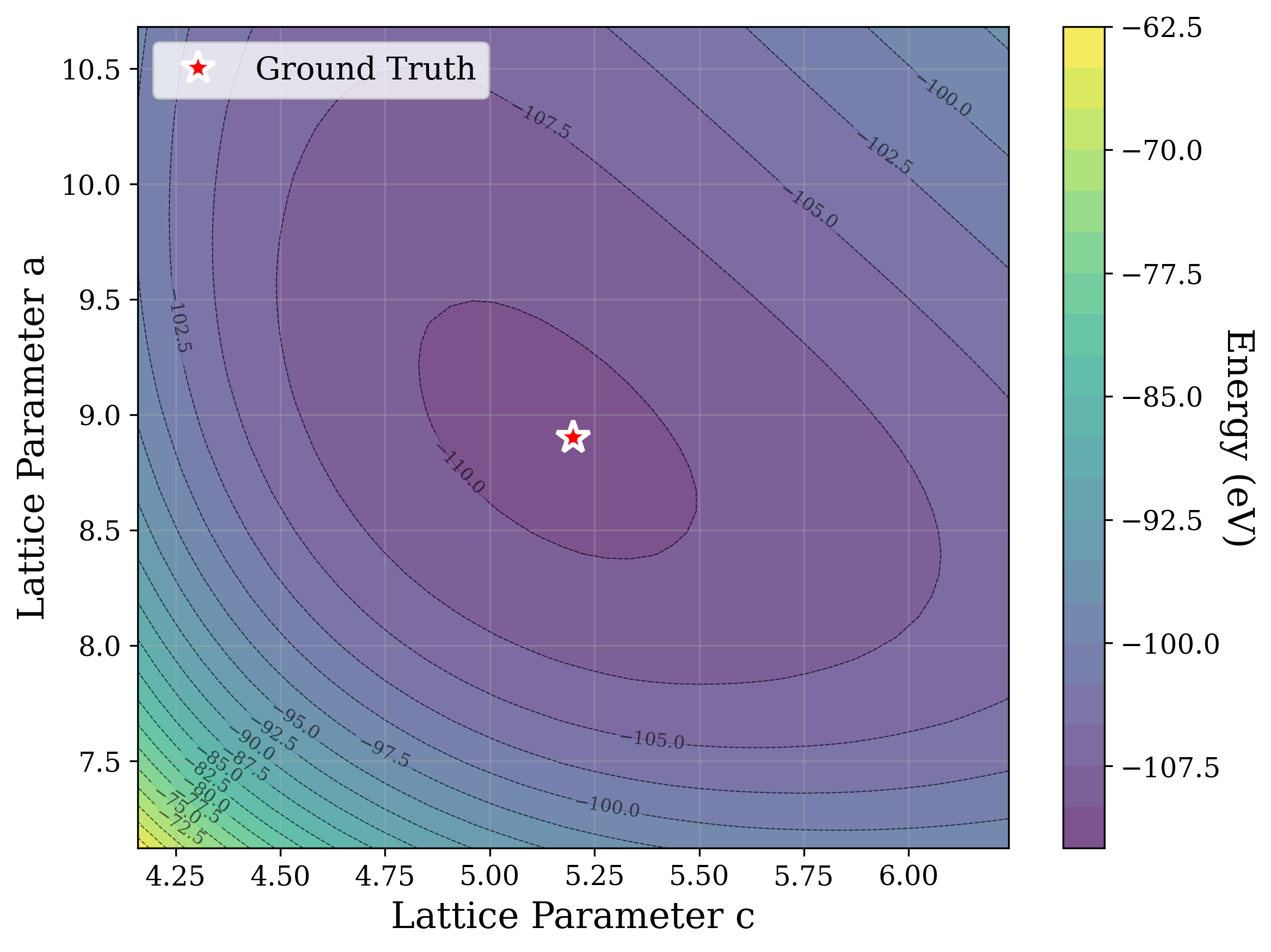
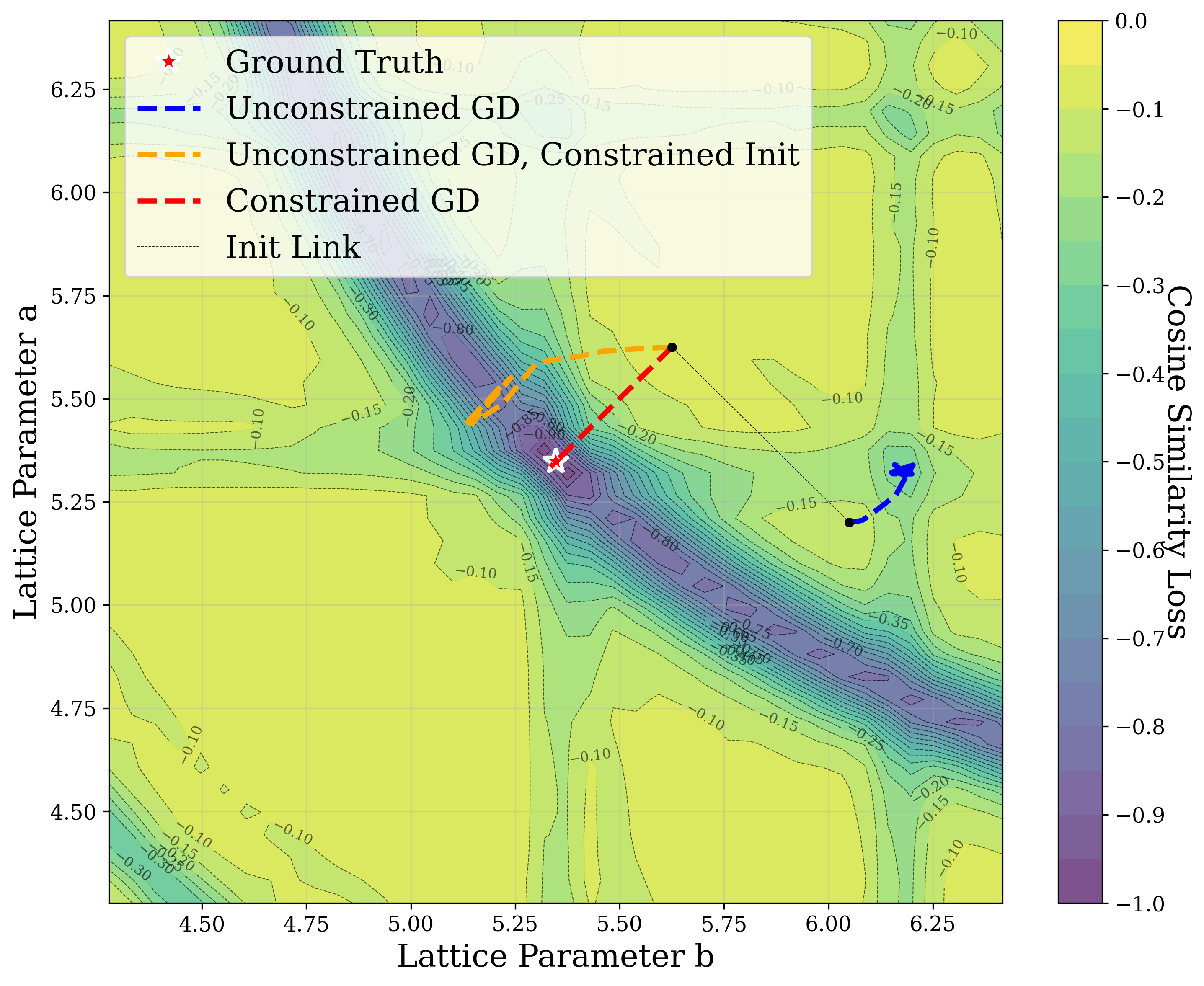
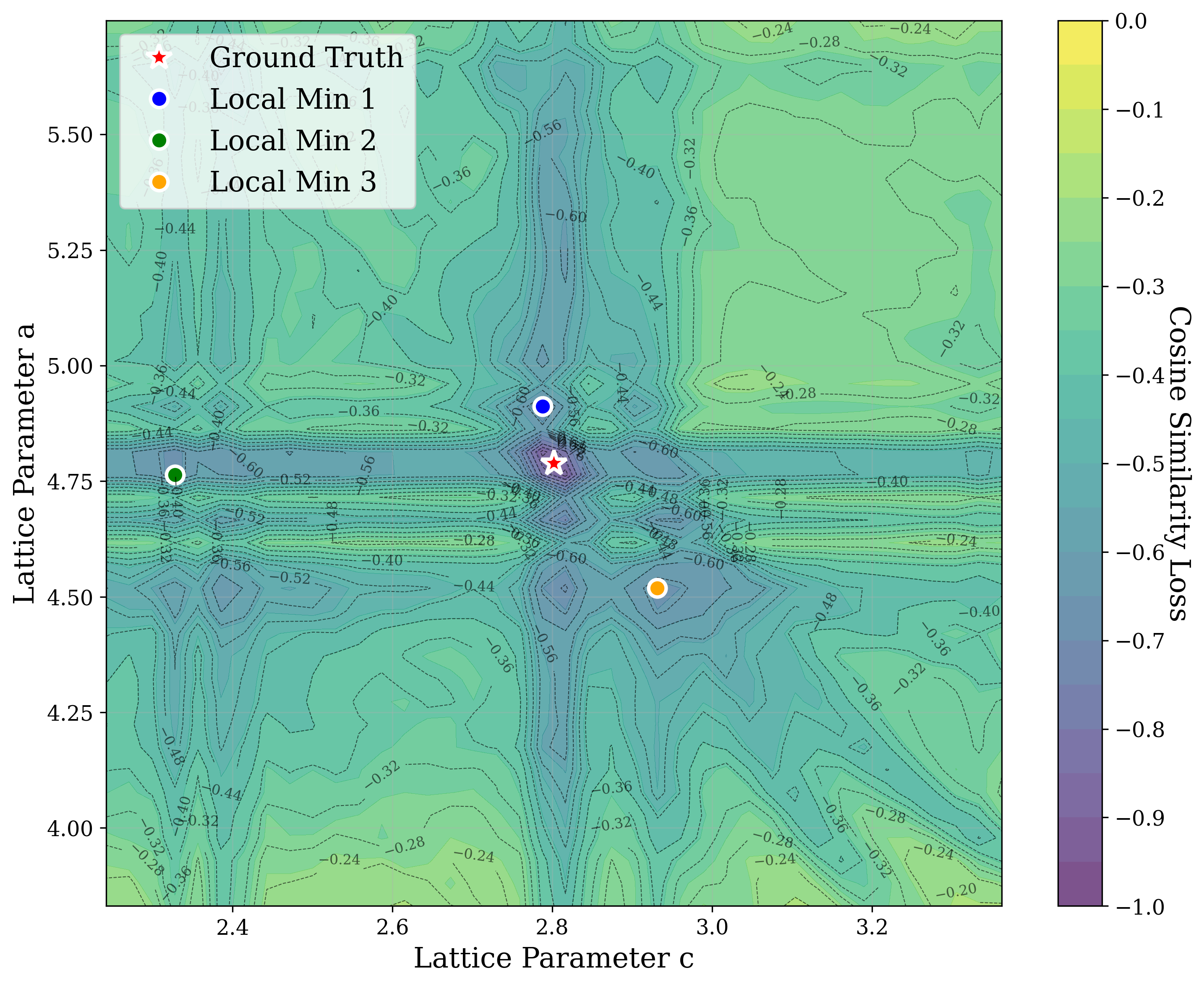
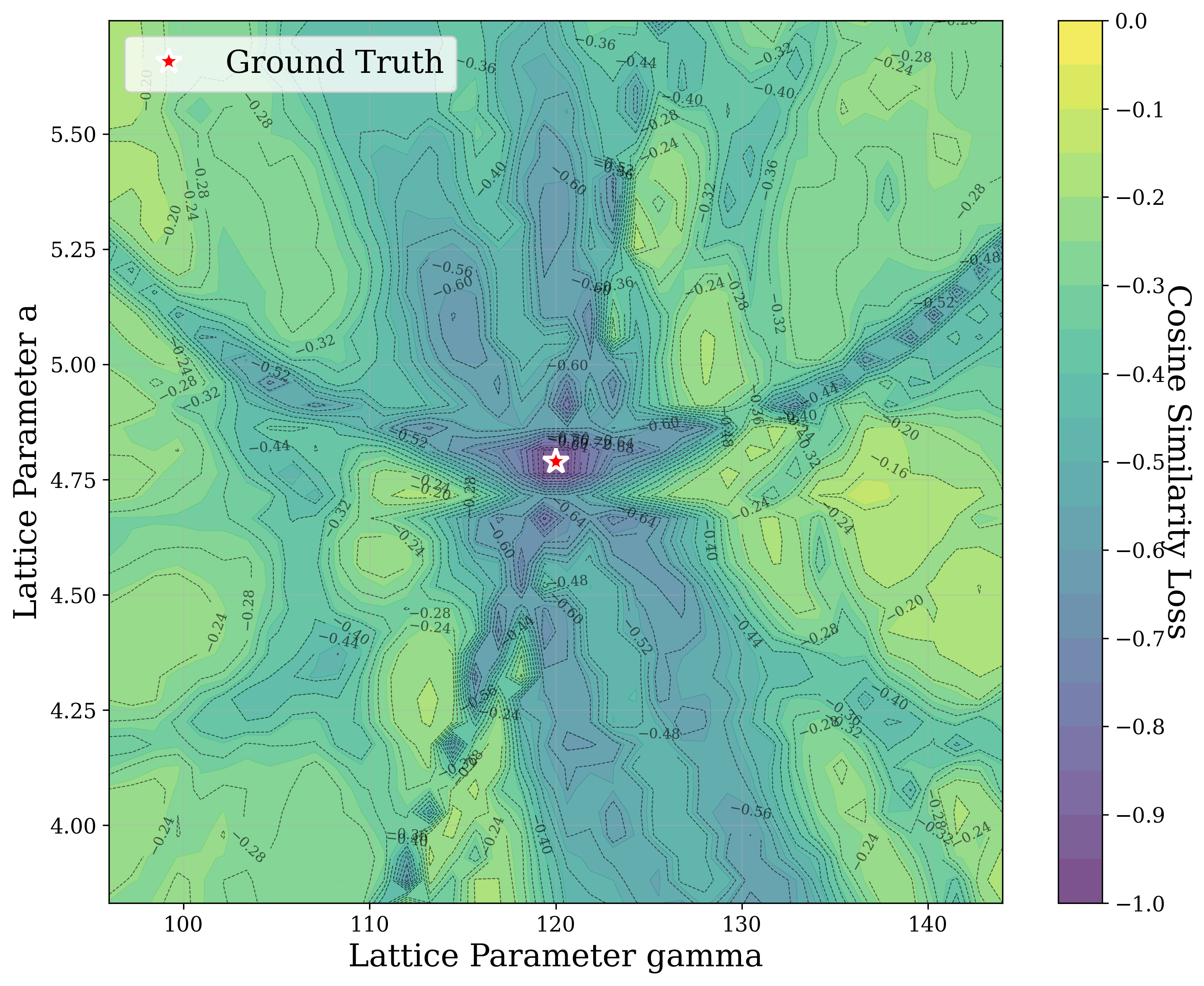
From a computational perspective, structural ambiguity remains even under idealized conditions. Two structures with different compositions can exhibit highly correlated XRD patterns if they share similar symmetry [15][16][17]. Moreover, even when stoichiometry is fixed, structures with close though distinct space groups can yield highly similar XRD patterns [2]. Notably, small distortions in lattice parameters or atomic coordinates can cause discontinuous changes in the diffraction pattern, such as the appearance or disappearance of peaks due to shifting Bragg conditions [4,Chapter 9]. This introduces a highly non-smooth relationship between structure and XRD signal.
Recently, there has been a surge of interest in crystal structure determination from XRD patterns using generative modeling [18][19][20][21][22][23]. A growing body of work applies gradient-based optimization approaches that leverage differentiable physics to refine generated or otherwise-obtained crystal structures by minimizing the difference between simulated and target XRD patterns. For example, Riesel et al. [19] generate crystals conditioned on a given XRD pattern and post-process them using a differentiable XRD simulator to update lattice parameters via gradient descent (GD). Parackal et al. [24] systematically enumerate candidate crystals given composition and space group inputs, and restricts the GD optimization to atomic positions along Wyckoff degrees of freedom. Lee et al. [25] create candidate crystals using an evolutionary algorithm, followed by crystals morphing by maximizing the cosine similarity between the XRD patterns. Outside the powder diffraction setting, GD has further been applied to determine lattice parameters from single-crystal diffraction patterns [26].
As gradient-based refinement relies on comparing simulated and target diffraction patterns, recent work has also focused on developing more robust XRD-similarity metrics. Otero-de-la Roza [27] introduced a cross-correlation-based metric that captures equivalence between diffraction patterns while remaining invariant to lattice distortions. Building on this work, Racioppi et al. [28] applied the metric to crystal structure prediction from XRD data, jointly optimizing the structure by minimizing both this similarity metric and the structure’s enthalpy. Hernández-Rivera et al. [29] systematically analyzed the sensitivity of different families of similarity metrics under isotropic lattice strain. Li et al. [30] proposed an entropy-based similarity measure for spectra and demonstrated its utility for molecular database retrieval from mass spectrometry data.
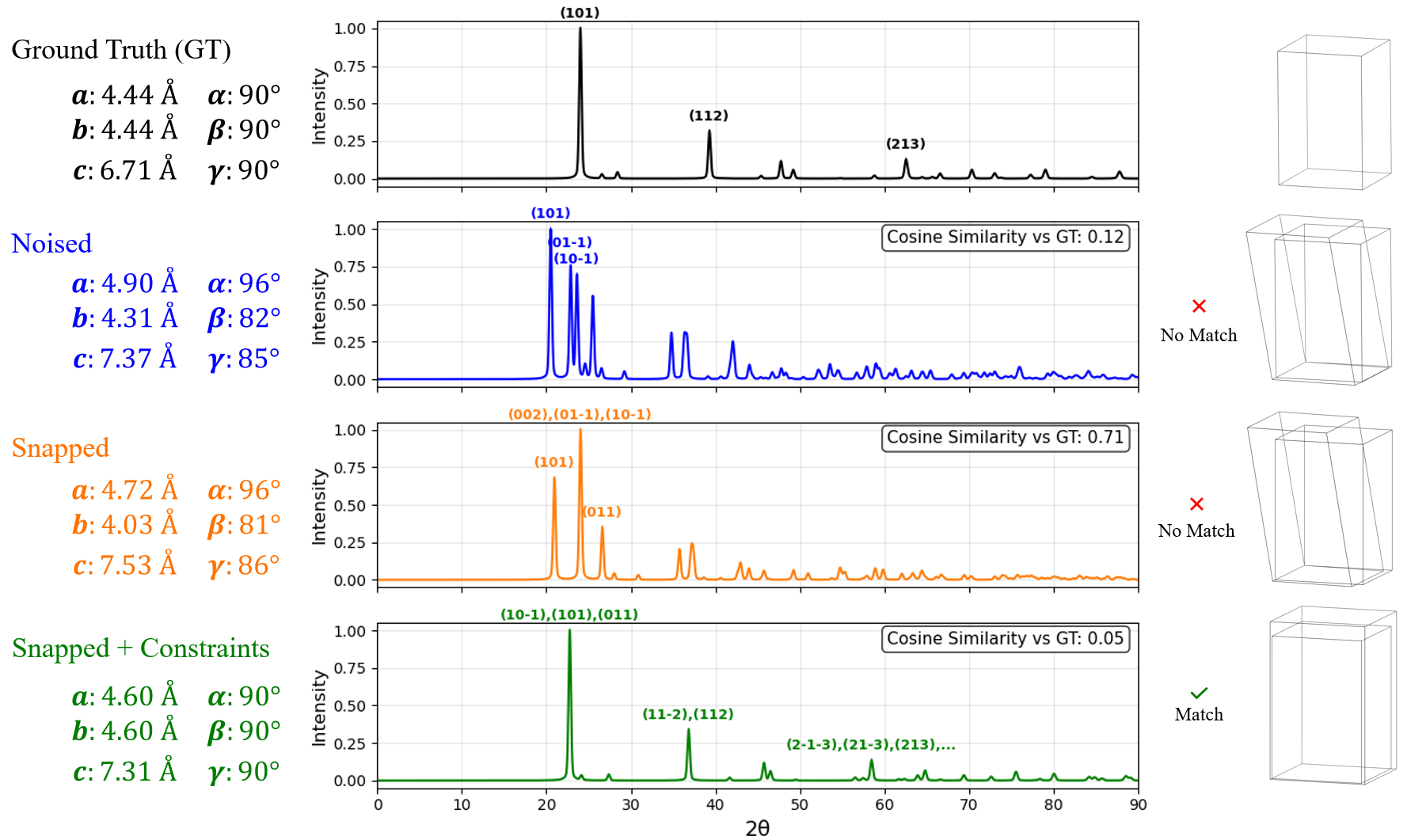
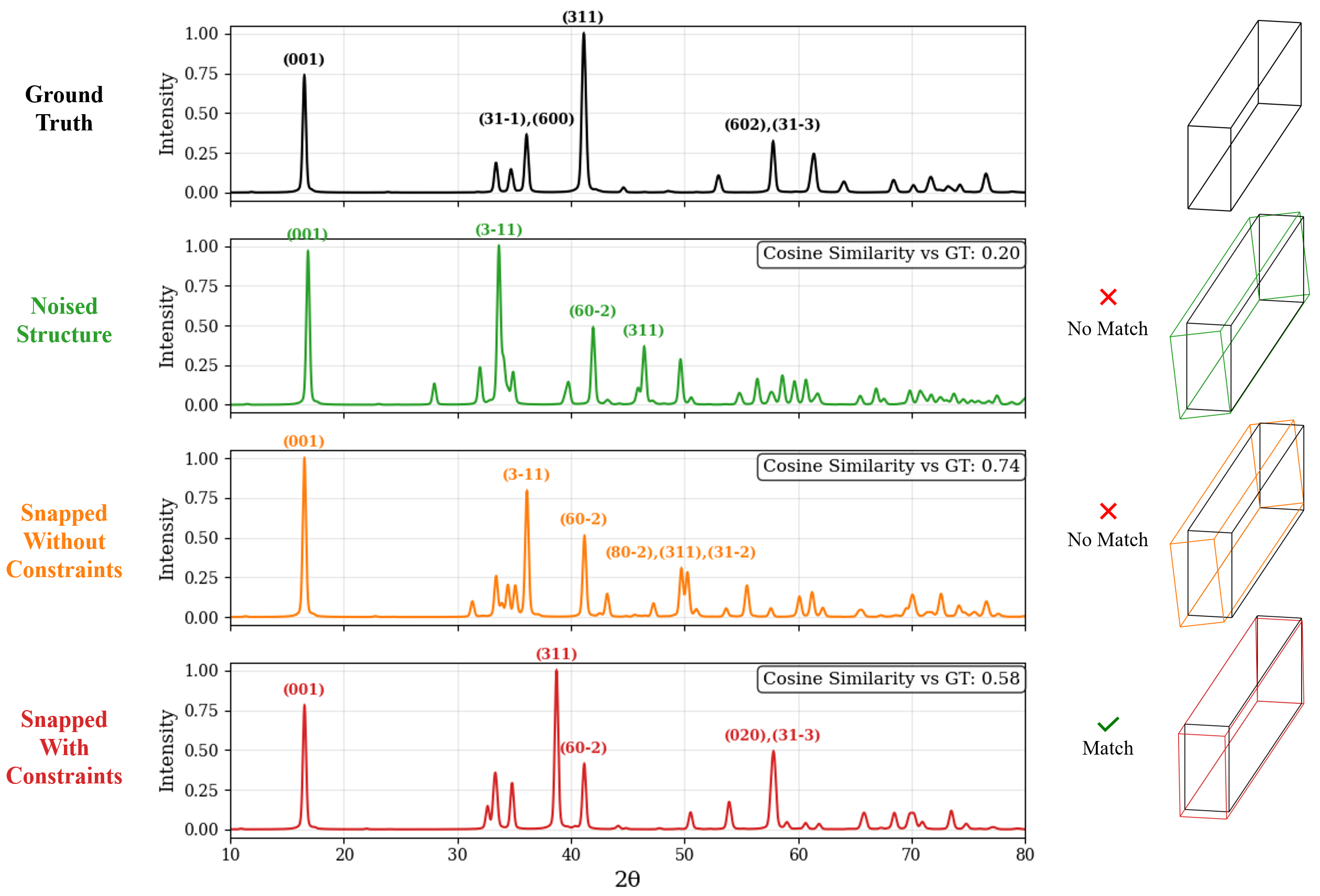
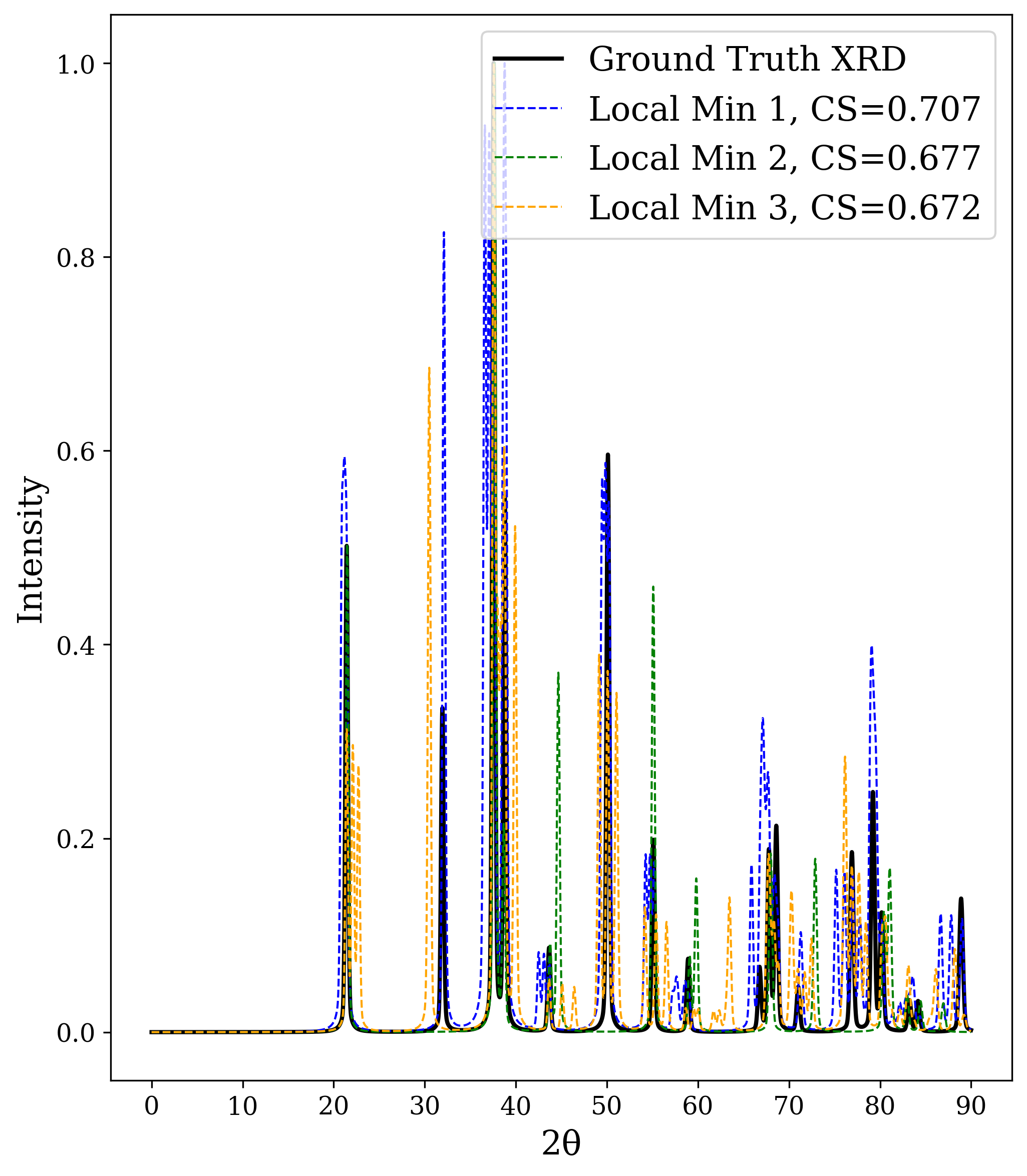
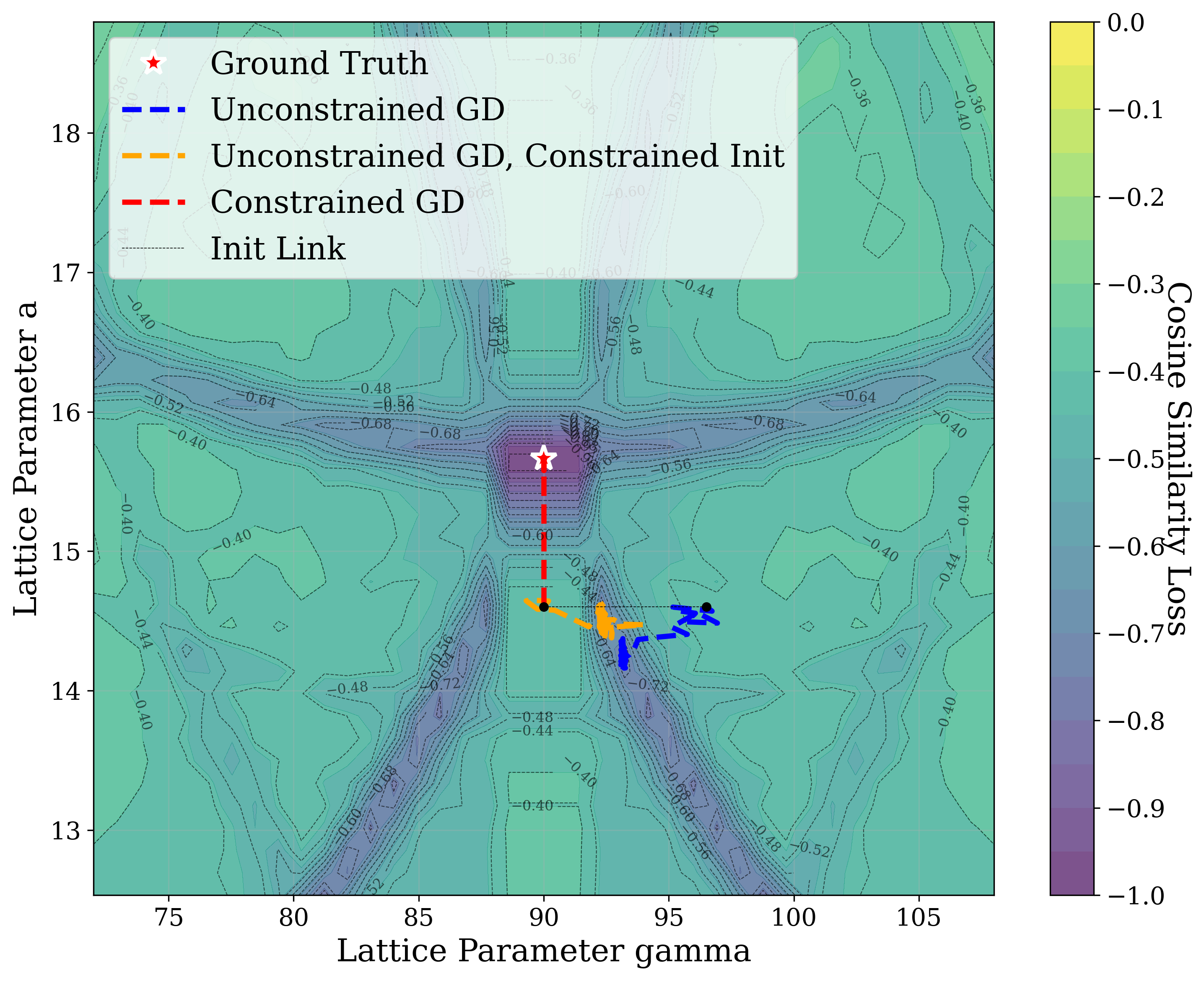
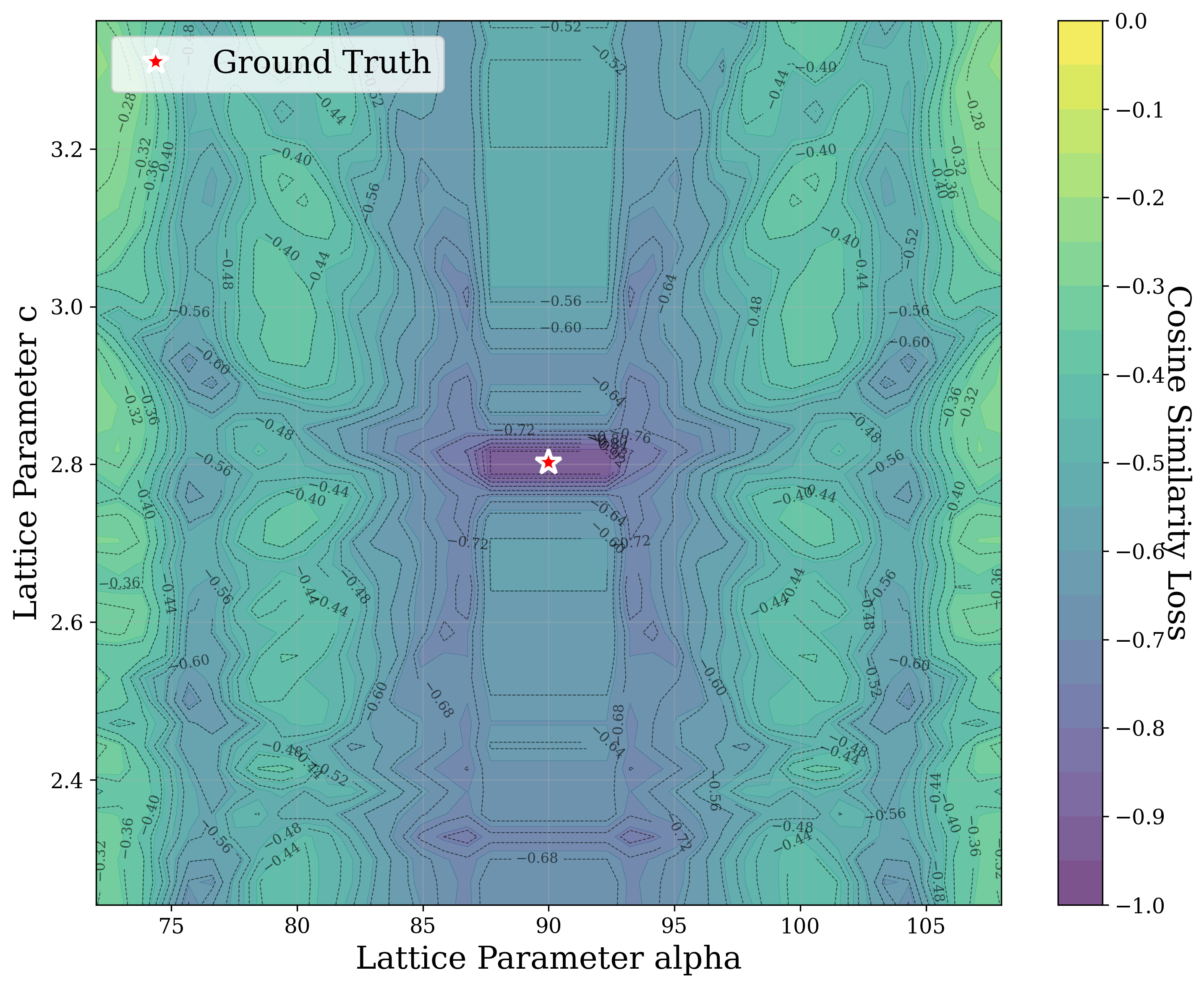
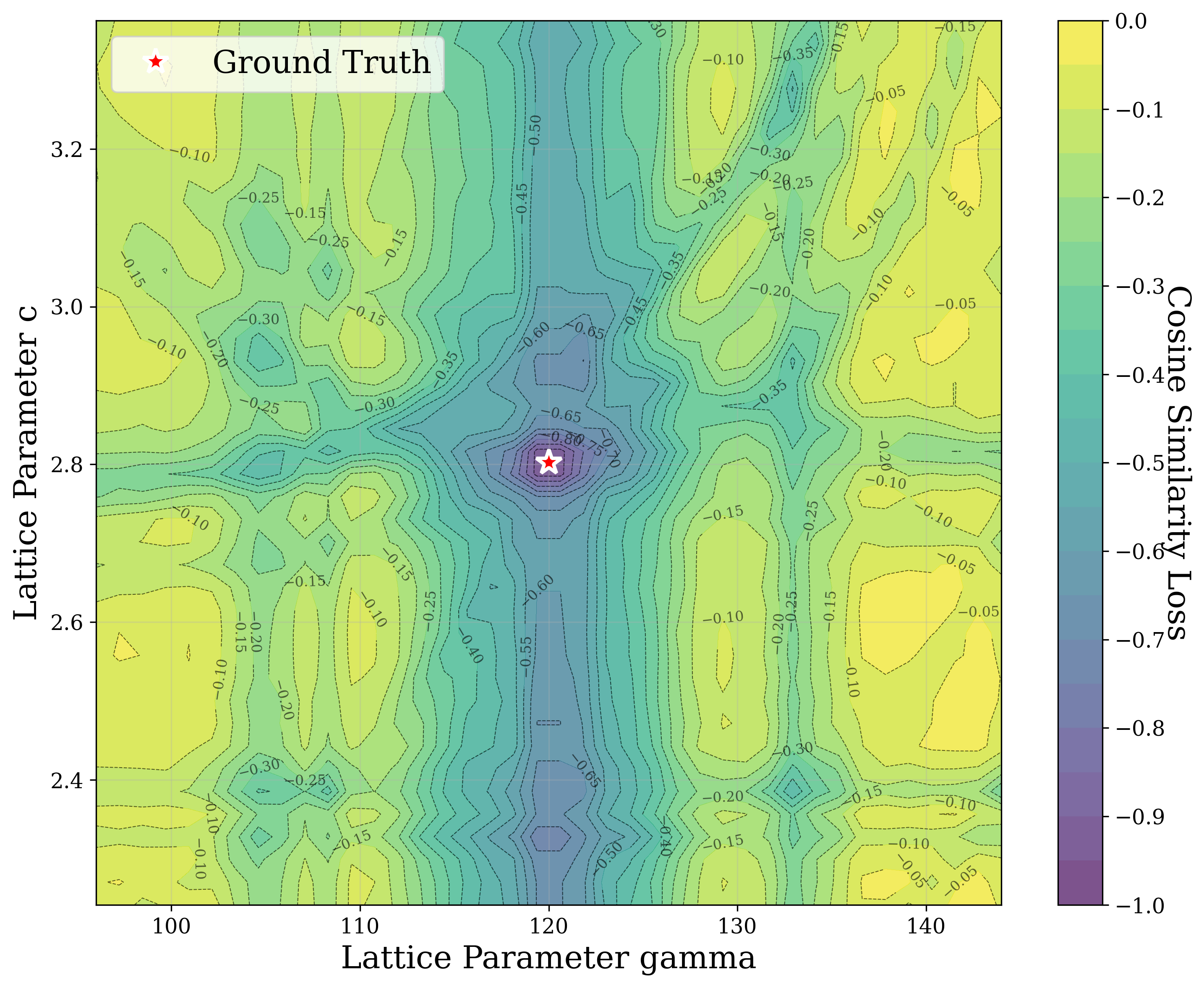
In this work, we explore the powder XRD-to-structure mapping through the lens of GD optimization. The goal is to recover correct structures based solely on XRD similarity from moderately deviated states. We ask whether the XRD landscape is locally smooth enough for GD to guide us back to the correct configuration. Inspired by experimentally observed symmetry-breaking effects such as thermal expansion from lattice vibrations and thermal fluctuations, we introduce two types of distortions: random lattice distortions and uncorrelated atomic displacements [8,[31][32][33]. These distortions resemble crystal structures predicted by generative models, which often produce nearly correct geometries but with imperfect symmetry [34][35][36]. Through this study, we examine the challenge of “the last mile” in structure elucidation from
…(Full text truncated)…
This content is AI-processed based on ArXiv data.