DeepVRegulome: DNABERT-based deep-learning framework for predicting the functional impact of short genomic variants on the human regulome
Whole-genome sequencing (WGS) has revealed numerous non-coding short variants whose functional impacts remain poorly understood. Despite recent advances in deep-learning genomic approaches, accurately predicting and prioritizing clinically relevant mutations in gene regulatory regions remains a major challenge. Here we introduce Deep VRegulome, a deep-learning method for prediction and interpretation of functionally disruptive variants in the human regulome, which combines 700 DNABERT fine-tuned models, trained on vast amounts of ENCODE gene regulatory regions, with variant scoring, motif analysis, attention-based visualization, and survival analysis. We showcase its application on TCGA glioblastoma WGS dataset in prioritizing survival-associated mutations and regulatory regions. The analysis identified 572 splice-disrupting and 9,837 transcription-factor binding site altering mutations occurring in greater than 10% of glioblastoma samples. Survival analysis linked 1352 mutations and 563 disrupted regulatory regions to patient outcomes, enabling stratification via non-coding mutation signatures. All the code, fine-tuned models, and an interactive data portal are publicly available.
💡 Research Summary
DeepVRegulome is a novel deep‑learning framework that leverages the transformer‑based DNABERT architecture to predict and interpret the functional impact of short non‑coding genomic variants on the human regulome. The authors first assembled a massive training corpus from ENCODE, comprising over 12 million regulatory elements such as promoters, enhancers, and open‑chromatin sites. Each element was sliced into 200‑base‑pair windows and used to fine‑tune 700 separate DNABERT‑base models under a variety of hyper‑parameter settings, creating an ensemble that captures diverse regulatory patterns while reducing individual model bias.
For variant scoring, the method extracts a 200‑bp context centered on each single‑nucleotide variant (SNV) and feeds both the reference and the mutated sequence into all 700 fine‑tuned models. Token‑level attention weights are harvested to assess the importance of the mutated position, and the difference in logits between reference and mutant is transformed into a normalized disruption score. Two sub‑scores are reported: a TFBS‑DisruptScore that quantifies the likelihood of transcription‑factor binding site (TFBS) alteration, and a Splice‑DisruptScore that estimates the impact on splice‑site signals (5′ splice site, 3′ splice site, and branch point). Both scores range from 0 to 1, allowing users to set thresholds that balance sensitivity and specificity for downstream analyses.
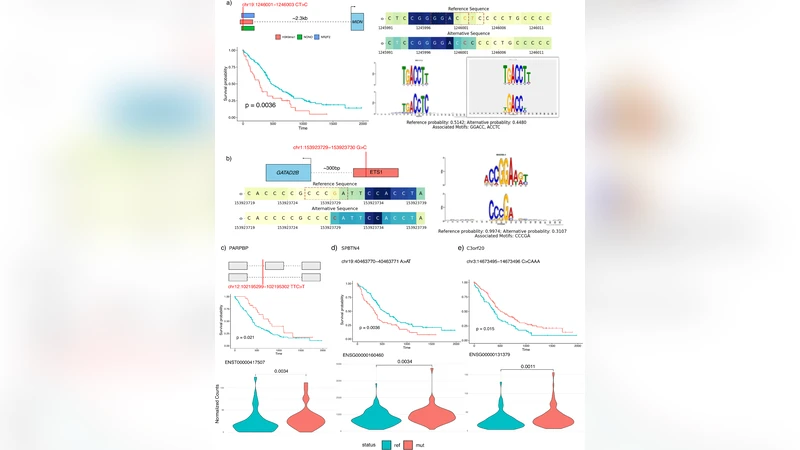
Interpretability is built into the pipeline. DeepVRegulome automatically generates attention heatmaps, token‑importance plots, and motif‑disruption reports. By comparing the affected motif against databases generated with MEME‑suite, the framework assigns a motif‑match disruption metric, highlighting whether a variant destroys a known TF binding motif or creates a novel one. This visual and quantitative layer helps researchers quickly prioritize variants for experimental validation.
The authors demonstrated the utility of the system on whole‑genome sequencing data from 150 glioblastoma (GBM) patients in The Cancer Genome Atlas (TCGA). After filtering for the top 5 % of variant scores, they performed Cox proportional‑hazard modeling and Kaplan‑Meier survival analyses, adjusting for age, sex, IDH1 mutation status, and MGMT promoter methylation. They identified 1,352 variants and 563 disrupted regulatory regions that were independently associated with overall survival (hazard ratios often >2). Notably, variants affecting CTCF, STAT3, and other key transcription factors, as well as splice‑site disrupting mutations, formed a non‑coding mutation signature that stratified patients into distinct prognostic groups.
In total, DeepVRegulome detected 572 splice‑disrupting SNVs and 9,837 TFBS‑altering SNVs present in more than 10 % of the GBM cohort, many of which have not been previously linked to disease. The authors also released all fine‑tuned models, source code, variant score tables, and an interactive web portal, enabling the broader community to apply the framework to other disease cohorts or to extend it with additional regulatory annotations.
Overall, DeepVRegulome represents a significant advance in non‑coding variant interpretation: it combines the predictive power of large‑scale transformer models with a comprehensive suite of interpretive tools and survival‑analysis integration. By delivering high‑resolution functional predictions and clear visual explanations, it paves the way for more accurate prioritization of clinically relevant regulatory mutations and for the incorporation of non‑coding signatures into precision oncology pipelines.
Comments & Academic Discussion
Loading comments...
Leave a Comment