Lithium Adsorbtion on Polyacenes $&$ Zig-zag-edge Graphene Strips
The effect of increased electron-density (from adsorbed Li atoms) in polyacenes and in nano-ribbons with zig-zag edge is discussed in terms of resonance theoretical considerations and in terms edge-localized frontier molecular orbitals. The argumentation from simple pictures is finally using the density functional theory (DFT) for anthracene, polyacene polymer and graphene strips. Some discussion is made for zig-zag edge graphene.
💡 Research Summary
The manuscript investigates how lithium atoms, when adsorbed onto polyacenes (with anthracene as the prototypical finite example) and onto graphene nanoribbons that possess zig‑zag edges, modify the electronic structure and geometry of these carbon‑based systems. Three complementary approaches are employed: (i) classical resonance theory, (ii) Hückel (tight‑binding) molecular‑orbital analysis, and (iii) density‑functional theory (DFT) calculations.
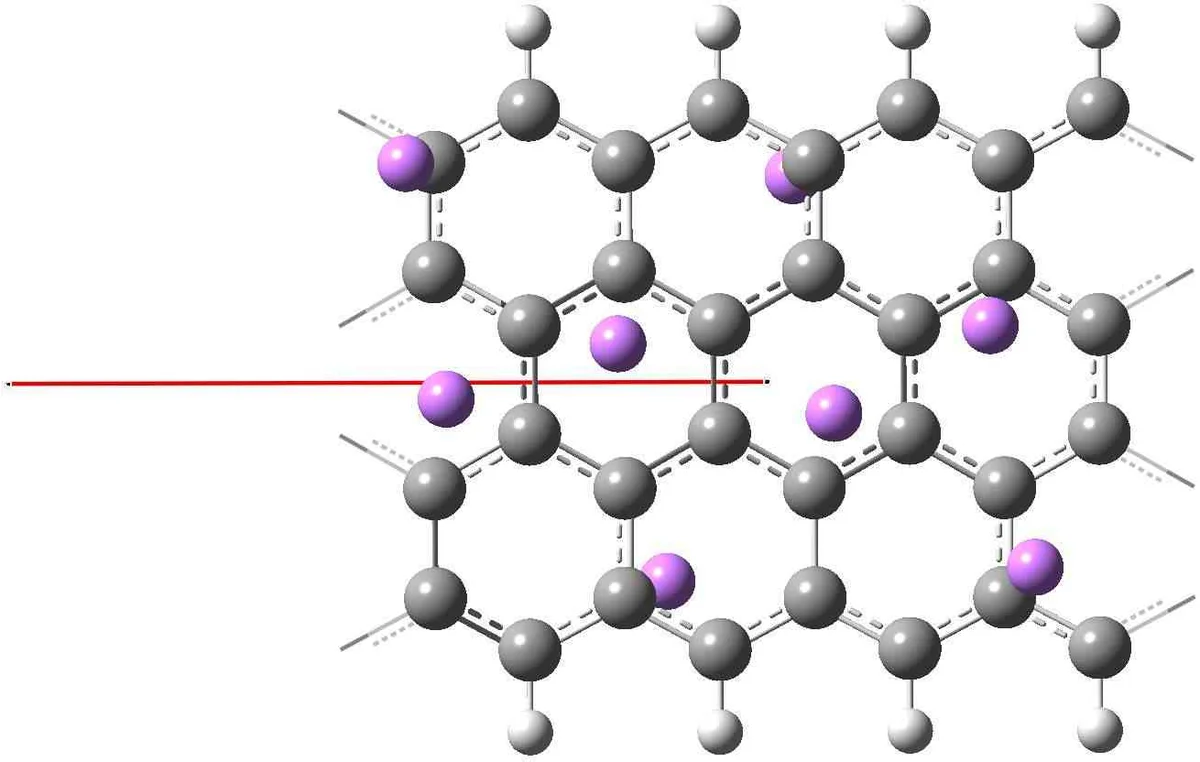
Resonance arguments, following Pauling and Wheland, show that lithium, being highly electropositive, donates its valence electron to the π‑network of the polyacene. The extra electron preferentially occupies the two carbon atoms located on the central symmetry plane of anthracene. This occupation weakens the central C=C bonds, allowing the terminal benzene‑like rings to adopt independent Clar sextet structures. Consequently, the central carbons become partially sp³‑hybridized, leading to a non‑planar distortion that is consistent with experimental observations of bent anthracene derivatives.
The Hückel model provides a quantitative counterpart. For an infinite polyacene chain the spectrum is symmetric about zero energy, and a non‑bonding (zero‑energy) orbital is localized on the “point” carbons, especially those near the chain centre. In finite polyacenes the LUMO retains this character, so electrons transferred from lithium populate orbitals that are concentrated on the same central sites identified by resonance theory. The model predicts a reduction of bond order for those C–C bonds, again foreshadowing sp³ rehybridization.
DFT calculations (B3LYP/6‑311G* and MP2/6‑311++G**) confirm both qualitative pictures. Optimized structures of anthracene with two lithium atoms placed on opposite sides of the molecular plane show almost complete charge transfer (Li charges ≈ +0.82 e and +0.87 e). The excess electron density accumulates on the two central carbons, which lengthen and tilt out of the plane, indicating a partial sp³ character. Natural Bond Orbital (NBO) analysis corroborates the ionic nature of the Li–C interaction. Similar periodic DFT studies on longer polyacene chains reveal a Peierls‑type distortion that doubles the unit cell, further supporting the resonance‑driven picture.
Extending the analysis to graphene, the authors treat semi‑infinite sheets with zig‑zag edges. Both resonance counting and tight‑binding theory predict a non‑bonding edge state that contributes 1/3 of an unpaired electron per edge unit cell. These edge‑localized states decay exponentially into the bulk, but different edge states have varying penetration depths, leading to substantial overlap and ferromagnetic coupling among the nominally unpaired spins. Lithium atoms preferentially bind to these edge sites because the edge LUMOs readily accept the donated electrons, resulting in strong Li–C binding, pronounced charge transfer, and local sp³ rehybridization of the edge carbons. In the interior of the sheet the same mechanism is weaker; lithium binds more weakly and at larger separations, consistent with the lower density of bulk Dirac‑cone states near the Fermi level.
The paper discusses practical implications for lithium‑ion batteries and 2‑D carbon functionalization. Lithium adsorption at edge or “point” carbons acts as n‑type doping, closing the intrinsic band gap of polyacenes and graphene nanoribbons, thereby enhancing electronic conductivity—a key factor for high‑rate battery electrodes. The near‑complete charge transfer and resulting sp³ distortion stabilize the lattice while preserving metallic character, especially when lithium atoms are arranged in paired top‑bottom configurations. The authors argue that the classical chemical concepts of Kekulé resonance, Clar sextets, and edge‑localized frontier orbitals remain powerful descriptors for modern nanomaterials, bridging molecular intuition with contemporary ab‑initio design strategies.
In summary, the work demonstrates a coherent, multi‑scale understanding of lithium adsorption on benzenoid carbon systems: resonance theory predicts where extra electrons will localize, Hückel theory quantifies the associated orbital character, and DFT validates the structural and energetic consequences. This integrated framework not only rationalizes existing experimental observations of Li‑decorated graphene but also provides guidance for engineering carbon‑based anodes with optimized lithium storage capacity and electronic performance.
Comments & Academic Discussion
Loading comments...
Leave a Comment