Investigating modularity in the analysis of process algebra models of biochemical systems
Compositionality is a key feature of process algebras which is often cited as one of their advantages as a modelling technique. It is certainly true that in biochemical systems, as in many other systems, model construction is made easier in a formalism which allows the problem to be tackled compositionally. In this paper we consider the extent to which the compositional structure which is inherent in process algebra models of biochemical systems can be exploited during model solution. In essence this means using the compositional structure to guide decomposed solution and analysis. Unfortunately the dynamic behaviour of biochemical systems exhibits strong interdependencies between the components of the model making decomposed solution a difficult task. Nevertheless we believe that if such decomposition based on process algebras could be established it would demonstrate substantial benefits for systems biology modelling. In this paper we present our preliminary investigations based on a case study of the pheromone pathway in yeast, modelling in the stochastic process algebra Bio-PEPA.
💡 Research Summary
The paper investigates how the inherent compositional structure of process‑algebra models, specifically those written in the stochastic process algebra Bio‑PEPA, can be exploited during the solution and analysis phase of biochemical system modeling. The authors begin by highlighting compositionality as a major advantage of process algebras: complex systems can be built from smaller, well‑defined components, making model construction more systematic and modular. However, they point out that the dynamic behavior of biochemical networks is characterized by strong inter‑dependencies, feedback loops, and non‑linear interactions, which pose serious challenges to any attempt at decomposed analysis.
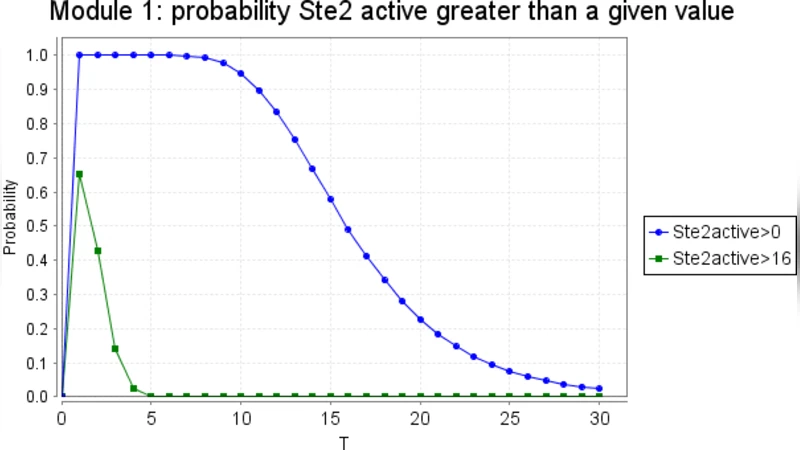
To explore these issues concretely, the authors use the yeast pheromone signaling pathway as a case study. This pathway involves ligand‑receptor binding, G‑protein activation, a MAPK cascade, and multiple positive and negative feedback mechanisms. Each biological entity (receptor, G‑protein, kinase, phosphatase, etc.) is encoded as a separate Bio‑PEPA process, and their interactions are expressed through synchronization actions such as “bind”, “activate”, and “phosphorylate”. The resulting model naturally forms a hierarchical tree, where each subtree can be interpreted as an independent Markov chain.
Two main decomposition strategies are examined. The first, static (structural) decomposition, partitions the full model into modules and replaces the influence of neighboring modules with interface parameters (e.g., average concentrations of input species). These parameters are either fixed a priori or estimated from preliminary simulations. Each module is then solved independently, and the results are combined to approximate the behavior of the whole system. While this approach theoretically reduces state‑space size, the authors find that inaccurate interface parameters quickly lead to cumulative errors because the biochemical system’s feedbacks cause rapid, context‑dependent changes in those parameters.
The second strategy, dynamic decomposition, monitors species concentrations during a stochastic simulation (implemented with Gillespie’s algorithm). When a species falls below a pre‑defined threshold, the entire module containing that species is temporarily disabled, effectively pruning low‑probability transitions from the state space. This adaptive reduction yields a roughly 30 % speed‑up in simulation time for the pheromone pathway without appreciable loss of accuracy in key observables such as final MAPK activation levels. Nevertheless, the method is highly sensitive to the choice of thresholds: overly aggressive pruning can eliminate rare but biologically critical events (e.g., switch‑like transitions) and can distort the dynamics when feedback loops become active.
From these experiments the authors draw several insights. First, the strong coupling among pathway components limits the feasibility of a fully modular solution; any decomposition must carefully account for cross‑module influences. Second, the quality of interface parameter estimation is the decisive factor for the success of static decomposition. Third, dynamic dimension reduction can improve computational efficiency, but it requires adaptive mechanisms that respect the system’s non‑linear response to perturbations.
In light of these findings, the paper proposes a hybrid decomposition framework for future work. Core feedback‑rich sub‑networks would be retained in the full model, while peripheral, loosely coupled modules would be analyzed separately and later integrated. The authors also suggest leveraging machine‑learning techniques to infer interface parameters on‑the‑fly and to adjust pruning thresholds adaptively based on real‑time error estimates.
Overall, the study demonstrates that while Bio‑PEPA’s formal compositionality greatly facilitates model construction, exploiting that compositionality for efficient solution of stochastic biochemical models is non‑trivial. Effective decomposition must balance computational gains against the risk of losing essential dynamical information, and it will likely require sophisticated, system‑aware strategies rather than naïve modular partitioning.
Comments & Academic Discussion
Loading comments...
Leave a Comment