COEM: Cross-Modal Embedding for MetaCell Identification
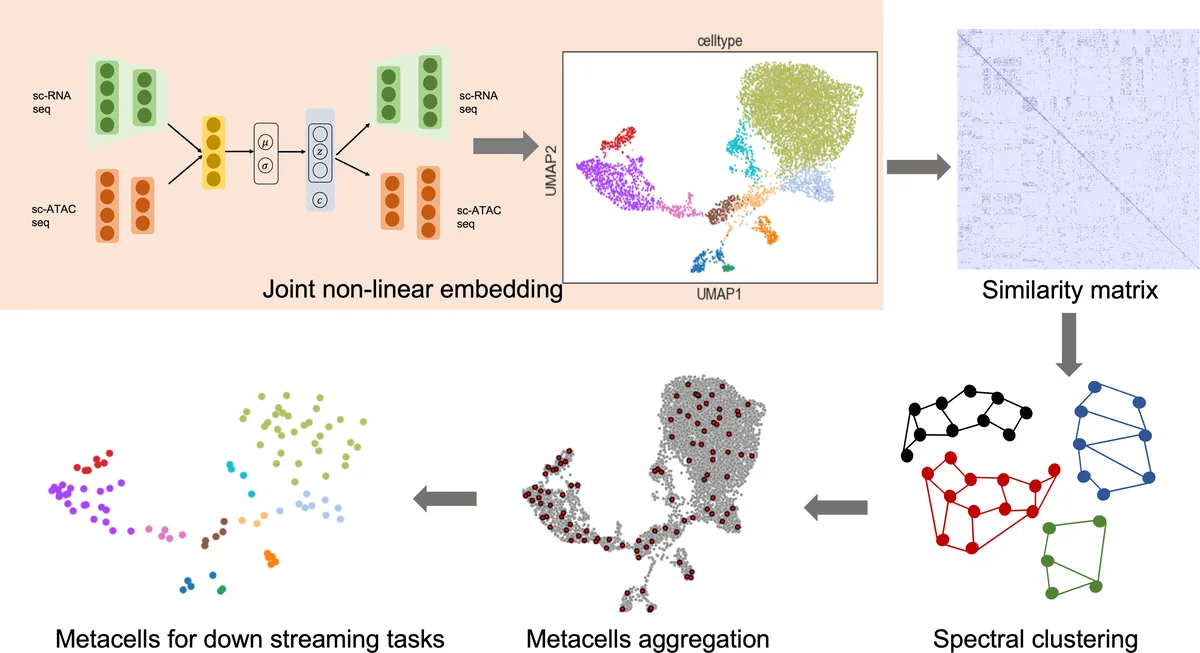
Metacells are disjoint and homogeneous groups of single-cell profiles, representing discrete and highly granular cell states. Existing metacell algorithms tend to use only one modality to infer metacells, even though single-cell multi-omics datasets profile multiple molecular modalities within the same cell. Here, we present \textbf{C}ross-M\textbf{O}dal \textbf{E}mbedding for \textbf{M}etaCell Identification (COEM), which utilizes an embedded space leveraging the information of both scATAC-seq and scRNA-seq to perform aggregation, balancing the trade-off between fine resolution and sufficient sequencing coverage. COEM outperforms the state-of-the-art method SEACells by efficiently identifying accurate and well-separated metacells across datasets with continuous and discrete cell types. Furthermore, COEM significantly improves peak-to-gene association analyses, and facilitates complex gene regulatory inference tasks.
💡 Research Summary
The paper introduces COEM (Cross‑Modal Embedding for MetaCell identification), a novel framework for constructing metacells from single‑cell multi‑omics data that simultaneously measures gene expression (scRNA‑seq) and chromatin accessibility (scATAC‑seq). Traditional metacell methods, such as MetaCell and SEACells, rely on a single modality, which limits their ability to capture the complex, often asynchronous relationship between transcription and chromatin state. Consequently, metacells derived from one modality can exhibit inflated negative peak‑to‑gene correlations, reflecting a “time‑lag” between chromatin opening and transcriptional activation.
COEM addresses this limitation by first learning a joint low‑dimensional representation of both modalities using a modified multi‑view variational auto‑encoder (VAE). The encoder consists of two attention‑based channels, one for scRNA‑seq and one for scATAC‑seq, whose outputs are concatenated to produce a posterior distribution qφ(z|x,y,c) over a shared latent variable z. The decoder mirrors this architecture to reconstruct both modalities. The generative model assumes a zero‑inflated Poisson distribution for gene counts, a negative‑binomial distribution for accessibility counts, and a Gaussian mixture prior for z conditioned on cell type c. This probabilistic formulation mitigates modality‑specific noise and aligns the two data types in a common space.
After obtaining the latent embeddings z, COEM builds a k‑nearest‑neighbors (KNN) graph based on Euclidean distances, then transforms the adjacency into a similarity matrix using a radial basis function (RBF) kernel to capture non‑linear relationships. Unlike SEACells, which employs archetypal analysis on the same embeddings, COEM applies spectral clustering to the similarity matrix. Spectral clustering leverages only graph connectivity, resulting in a computationally cheaper operation (approximately an order of magnitude faster in the reported benchmarks) while preserving the ability to delineate fine‑grained cellular states.
The authors evaluate COEM on five publicly available multi‑omics datasets: sci‑CAR cell lines, SNARE‑seq cell lines, 10X Genomics Multiome PBMCs, CD34⁺ bone‑marrow progenitors, and T‑cell‑depleted bone‑marrow samples. Three quantitative metrics are used: compactness (within‑metacell homogeneity), separation (between‑metacell distinctness), and cell‑type purity (fraction of cells from the same annotated type within a metacell). Across most datasets, COEM achieves higher purity and separation than SEACells‑ATAC and SEACells‑RNA, while maintaining comparable compactness. The advantage is especially pronounced in datasets with continuous developmental trajectories, where COEM’s joint embedding captures subtle transitional states that single‑modality methods miss.
A key biological validation concerns peak‑to‑gene correlation analysis. For each metacell, the authors compute Pearson correlations between gene expression and accessibility of peaks located within ±100 kb of the gene. In SEACells‑ATAC metacells, 23–25 % of peak‑gene pairs show negative correlations, reflecting the time‑lag artifact. In contrast, COEM metacells exhibit only 1–3 % negative correlations, aligning with previously reported single‑cell estimates (≈1–11 %). Moreover, core regulatory genes such as GATA2 display stronger positive correlations in COEM metacells (e.g., 0.73 vs. 0.62 in SEACells‑ATAC), indicating more accurate regulatory inference.
The discussion emphasizes that integrating scRNA‑seq and scATAC‑seq via a joint VAE yields a latent space that faithfully represents cellular identity, enabling more reliable metacell construction. COEM’s faster runtime and reduced bias in peak‑to‑gene links make it attractive for downstream tasks such as cis‑regulatory element (CRE) prediction, gene‑regulatory network reconstruction, and multi‑omics velocity modeling. The authors propose future extensions, including causally informed representation learning, optimal‑transport based modeling of metacell transitions across modalities, and broader benchmarking on emerging multi‑omics platforms. In summary, COEM demonstrates that cross‑modal embedding combined with spectral clustering substantially improves metacell quality and downstream regulatory analyses compared to existing single‑modality approaches.
Comments & Academic Discussion
Loading comments...
Leave a Comment