A comparative study of Gaussian Graphical Model approaches for genomic data
The inference of networks of dependencies by Gaussian Graphical models on high-throughput data is an open issue in modern molecular biology. In this paper we provide a comparative study of three methods to obtain small sample and high dimension estimates of partial correlation coefficients: the Moore-Penrose pseudoinverse (PINV), residual correlation (RCM) and covariance-regularized method $(\ell_{2C})$. We first compare them on simulated datasets and we find that PINV is less stable in terms of AUC performance when the number of variables changes. The two regularized methods have comparable performances but $\ell_{2C}$ is much faster than RCM. Finally, we present the results of an application of $\ell_{2C}$ for the inference of a gene network for isoprenoid biosynthesis pathways in Arabidopsis thaliana.
💡 Research Summary
The paper addresses the challenge of estimating partial correlation networks from high‑throughput genomic data, where the number of variables (genes) far exceeds the number of samples (n < p). In Gaussian Graphical Models (GGMs), conditional independence between two variables is encoded by the partial correlation coefficient, which can be derived from the precision matrix Θ = Σ⁻¹. When n > p, Θ can be obtained by directly inverting the sample covariance matrix Σ, but in the typical genomic setting (n < p) Σ is singular and regularization is required.
Three estimation strategies are compared:
-
Moore‑Penrose Pseudoinverse (PINV) – The sample covariance matrix S is decomposed by singular value decomposition (SVD) and its Moore‑Penrose pseudoinverse S⁺ is used as an estimate of Θ. This approach is straightforward but suffers from numerical instability when small singular values are inverted, especially when p is close to n.
-
Residual Correlation Method (RCM) – For each pair of variables (i, j) a regularized least‑squares (ridge) regression is performed on the remaining p‑2 variables. The residuals r_i and r_j are then correlated, yielding an estimate of the partial correlation. The ridge penalty λ is selected by leave‑one‑out cross‑validation. While theoretically sound, RCM requires solving O(p²) ridge regressions, making it computationally demanding.
-
ℓ₂‑Regularized Covariance Method (ℓ₂C) – The log‑likelihood of a multivariate normal model is penalized with an ℓ₂ (Frobenius‑norm) term λ‖Θ‖_F². The optimality condition Θ⁻¹ − 2λΘ = S can be rewritten as an eigenvalue problem. If s_i are the eigenvalues of S, the corresponding eigenvalues of Θ are given analytically by θ_i = (‑s_i + √(s_i² + 8λ))/4λ. Thus, after a single eigen‑decomposition of S, the entire precision matrix is obtained in closed form. The penalty λ is tuned by 20 random train/validation splits, maximizing the penalized log‑likelihood on the validation set.
Simulation Study
Datasets were generated for p = 50, 200, 400 and n = 20, 200, 500 under three network topologies: random, hub‑centric, and clique. For each configuration 20 replicates were created. Performance was measured by the area under the ROC curve (AUC) for edge detection and by wall‑clock time.
- When n > p, all three methods achieved AUC ≈ 1, confirming that regularization is unnecessary in this regime.
- In the n < p regime, PINV displayed marked instability, especially when p ≈ n, a phenomenon the authors refer to as a “resonance effect” caused by the amplification of noise through small singular values.
- Both ℓ₂C and RCM maintained high AUC (≥ 0.95) across all topologies and sample sizes. In the random topology with n = 20 and p = 200 or 400, RCM’s AUC was about 10 % higher than ℓ₂C’s.
- Timing results highlighted a stark contrast: ℓ₂C required only a few hundredths of a second (≈ 0.03 s) per dataset, whereas RCM needed roughly 0.8–1.0 s, reflecting the cost of solving many ridge regressions. PINV was fast but its AUC variability made it unreliable.
Application to Arabidopsis Isoprenoid Pathways
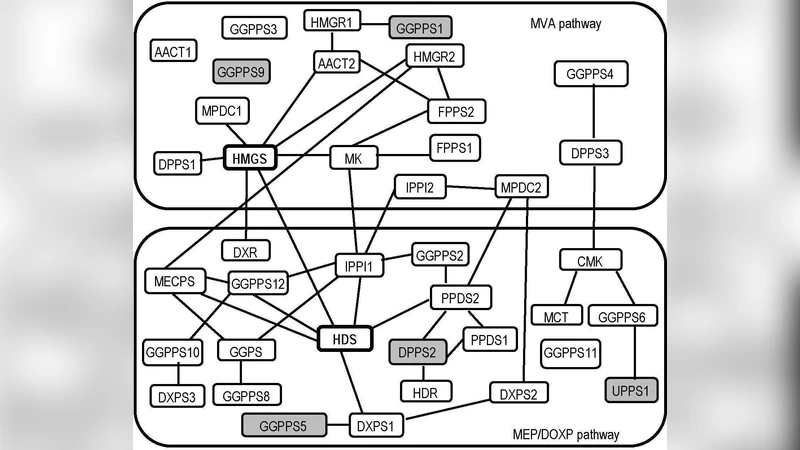
The authors applied ℓ₂C to a real microarray dataset comprising expression measurements for 39 genes directly involved in the two isoprenoid biosynthetic routes (the cytosolic mevalonate (MV A) pathway and the plastidial MEP/DOXP pathway) and 795 genes from 56 other metabolic pathways, across 118 samples. A bootstrap procedure generated 95 % confidence intervals for each estimated partial correlation; an edge was retained only if zero lay outside this interval. The resulting network contained 44 edges, forming two densely connected modules corresponding to the two pathways. Notably, two genes—HMGS (hydroxymethylglutaryl‑CoA synthase) and HDS (hydroxymethyl‑butenyl diphosphate synthase)—emerged as strong candidates mediating cross‑talk between the pathways, supporting earlier biochemical evidence of metabolic interaction.
Conclusions
The study demonstrates that ℓ₂C offers a compelling balance of statistical accuracy and computational efficiency for high‑dimensional GGM inference. Its closed‑form solution via eigen‑decomposition makes it scalable to thousands of variables, a crucial advantage over RCM, whose accuracy is comparable but whose runtime is prohibitive in large‑p settings. PINV, while simple to implement, is vulnerable to instability when the sample size approaches the number of variables, limiting its practical utility. Overall, the paper provides a clear, data‑driven recommendation: for n < p genomic analyses, ℓ₂‑regularized covariance estimation should be the method of choice, with RCM reserved for situations where its slightly higher AUC justifies the extra computational cost.
Comments & Academic Discussion
Loading comments...
Leave a Comment