Identification of cromosomal translocation hotspots via scan statistics
The detection of genomic regions unusually rich in a given pattern is an important undertaking in the analysis of next generation sequencing data. Recent studies of chromosomal translocations in activated B lymphocytes have identified regions that are frequently translocated to c-myc oncogene. A quantitative method for the identification of translocation hotspots was crucial to this study. Here we improve this analysis by using a simple probabilistic model and the framework provided by scan statistics to define the number and location of translocation breakpoint hotspots. A key feature of our method is that it provides a global chromosome-wide significance level to clustering, as opposed to previous methods based on local criteria. Whilst being motivated by a specific application, the detection of unusual clusters is a widespread problem in bioinformatics. We expect our method to be useful in the analysis of data from other experimental approaches such as of ChIP-seq and 4C-seq.
💡 Research Summary
The paper addresses the problem of detecting genomic regions that contain an unusually high density of chromosomal translocation breakpoints, a task that is central to the analysis of next‑generation sequencing (NGS) data from activated B lymphocytes. Existing approaches, such as those used in previous studies (e.g., the negative‑binomial based “local” method), define clusters by concatenating nearby breakpoints and assess significance with a test that treats each cluster independently. While useful, these methods provide only local p‑values and do not control the overall false‑positive rate across an entire chromosome, limiting their ability to compare hotspot size and frequency under different biological conditions.
To overcome this limitation, the authors introduce a scan‑statistic framework rooted in classical spatial statistics. They model the occurrence of breakpoints along a chromosome of length N as a realization of an independent and identically distributed Bernoulli process with success probability p = a/N, where a is the total number of observed breakpoints. For a fixed window width m (e.g., 500 bp or 5 kbp), they compute the number of successes Y_i in each sliding window i and define the scan statistic S_m = max_i Y_i, i.e., the maximum count observed in any window. The key statistical question is the tail probability P(S_m ≥ y) under the null hypothesis of random placement. Exact calculation would involve a hypergeometric distribution, but because N is typically >10⁶ and m is at least several hundred bases, the authors adopt an asymptotic approximation that replaces the hypergeometric with a binomial distribution. They further simplify the binomial tail using a closed‑form expression involving the Gauss hypergeometric function ₂F₁, yielding a fast and accurate p‑value formula (Equation 3).
Having a global p‑value for each window, the authors perform a chromosome‑wide scan: each window i is tested against a null hypothesis H₀,i, and windows with raw p‑values ≤ α_H (set to 0.05) are collected into a set B. Because adjacent windows overlap, the resulting tests are positively dependent. To control the false discovery rate (FDR) under dependence, they apply the Benjamini‑Yekutieli correction, producing adjusted p‑values p₁,…,p_{N‑m+1}. Windows that survive the correction (p* ≤ α_H) form a refined set B*. Connected components of B* are merged, trimmed to the exact breakpoint boundaries, and finally evaluated for significance based on their length ℓ using the same scan‑statistic formula with θ = ℓ/N. The final collection of significant regions is denoted B† and constitutes the identified translocation hotspots.
The authors implement this procedure in an open‑source tool called hot_scan, written in Perl and R, leveraging Parallel::ForkManager for multicore execution and the GNU Scientific Library (via Math::GSL::SF) for hypergeometric function evaluation. The software is freely available on GitHub.
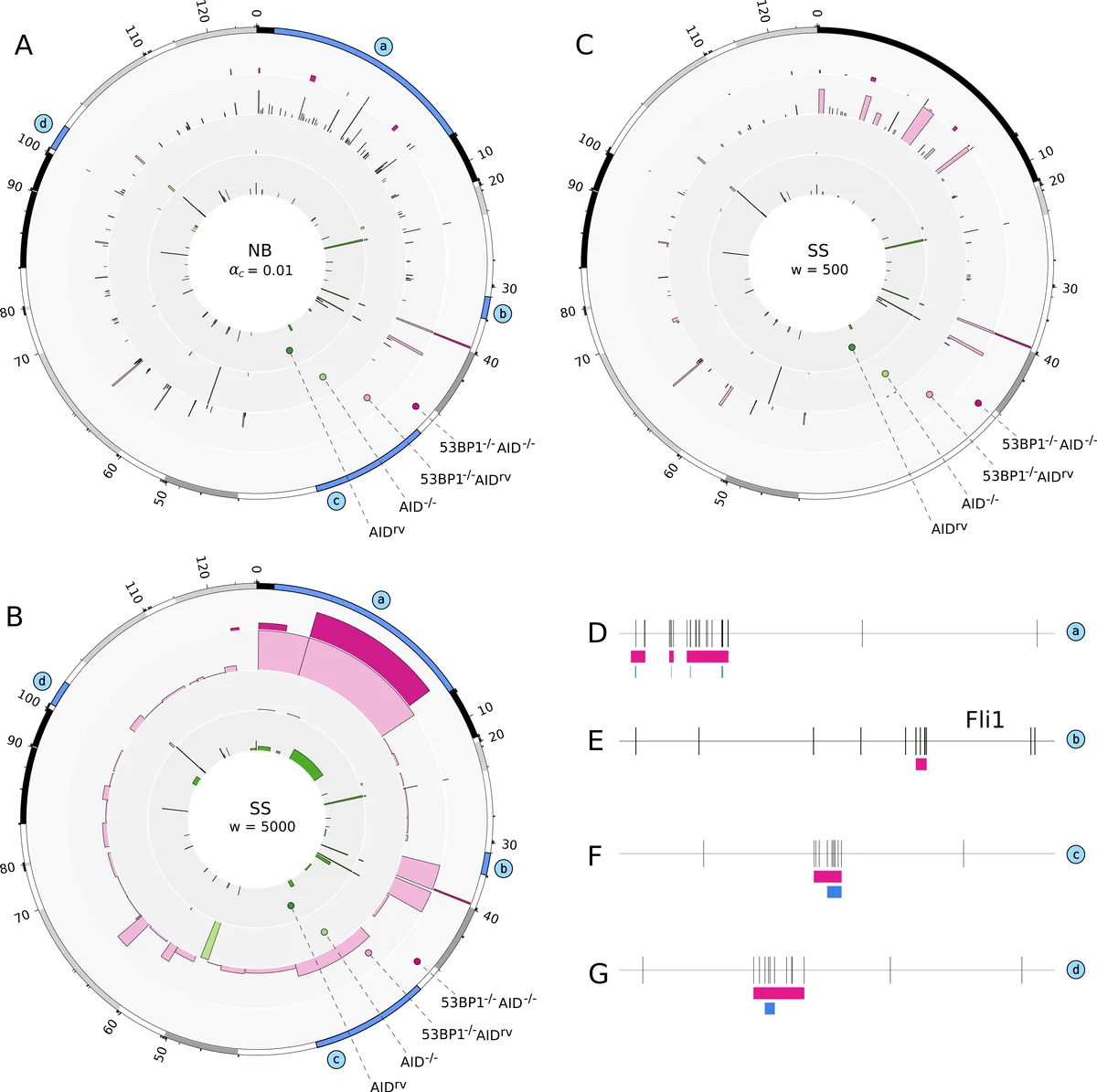
To benchmark the new method, the authors re‑analyze four previously published TC‑Seq datasets representing different B‑cell conditions: (1) AID‑expressing activated B cells, (2) AID‑knockout cells, (3) 53BP1‑deficient cells expressing AID, and (4) 53BP1‑deficient cells lacking AID. They compare the scan‑statistic based hotspots (SS₅₀₀ and SS₅₀₀₀) with those obtained using the earlier local negative‑binomial approach (NB₀.₀₁ and NB₀.₀₅). The scan‑statistic consistently identifies longer hotspots than the local method, especially in the 53BP1‑deficient background where DNA end resection is enhanced. When AID is over‑expressed in the 53BP1‑deficient context, hotspot lengths increase further, a pattern that the local method fails to capture because its significance threshold is applied to individual inter‑breakpoint distances rather than to the global clustering pattern.
Biologically, several of the newly discovered hotspots overlap known tumor‑suppressor genes, suggesting that aberrant translocations may target these loci. Integration with Pol II ChIP‑seq data shows that many hotspots coincide with regions of high transcriptional activity, supporting the hypothesis that transcription‑associated DNA damage contributes to translocation formation. Functional enrichment analysis using WebGestalt reveals that genes within scan‑statistic hotspots are significantly over‑represented in pathways related to DNA repair, immune response, and cell cycle regulation.
In summary, the paper makes the following contributions:
- Methodological Innovation – Introduction of a scan‑statistic based global test for breakpoint clustering, providing chromosome‑wide control of false positives.
- Statistical Derivation – Development of a practical p‑value approximation using binomial and hypergeometric asymptotics, validated for large‑scale genomic data.
- Multiple‑Testing Adjustment – Application of the Benjamini‑Yekutieli procedure to account for dependence among overlapping windows.
- Software Release – An open‑source, parallelizable implementation (hot_scan) that can be applied to any genomic coordinate dataset.
- Biological Insight – Demonstration that hotspot size and frequency are modulated by DNA repair status (53BP1) and AID activity, uncovering longer, previously hidden hotspots that overlap oncogenic and tumor‑suppressor loci.
- Generalizability – The framework is applicable beyond translocation data, potentially serving ChIP‑seq, 4C‑seq, Hi‑C, or any assay where spatial clustering of reads is of interest.
Overall, the study provides a robust statistical tool for the detection of genomic hotspots, improves upon existing local methods, and yields new biological hypotheses about the mechanisms driving chromosomal translocations in B cells.
Comments & Academic Discussion
Loading comments...
Leave a Comment