A complete data processing workflow for CryoET and subtomogram averaging
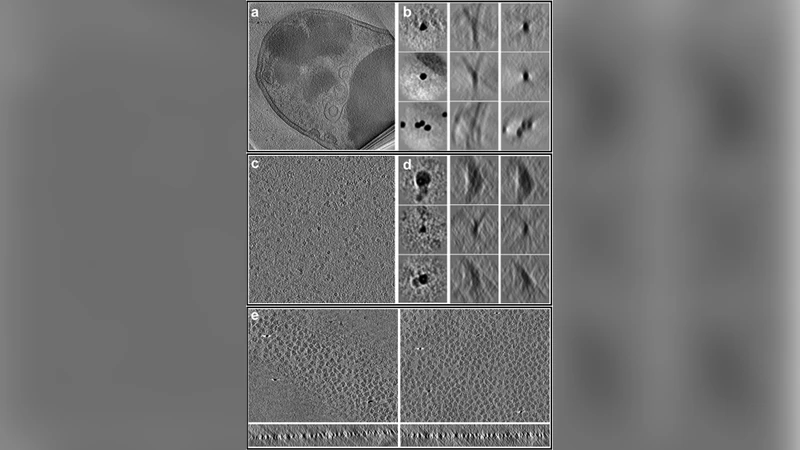
Electron cryotomography (CryoET) is currently the only method capable of visualizing cells in 3D at nanometer resolutions. While modern instruments produce massive amounts of tomography data containing extremely rich structural information, the data processing is very labor intensive and results are often limited by the skills of the personnel rather than the data. We present an integrated workflow that covers the entire tomography data processing pipeline, from automated tilt series alignment to subnanometer resolution subtomogram averaging. This workflow greatly reduces human effort and increases throughput, and is capable of determining protein structures at state-of-the-art resolutions for both purified macromolecules and cells.
💡 Research Summary
The paper presents a fully integrated, largely automated workflow that spans the entire cryo‑electron tomography (cryo‑ET) data‑processing pipeline, from tilt‑series alignment to sub‑nanometer subtomogram averaging. The authors begin by highlighting the current bottleneck: modern cryo‑ET instruments generate terabytes of data per experiment, yet the downstream processing remains labor‑intensive, highly dependent on expert knowledge, and consequently limits the achievable resolution. To address this, they designed a modular software stack that links all major processing steps—tilt‑series alignment, motion correction, contrast‑transfer‑function (CTF) estimation, particle picking, and subtomogram alignment/averaging—into a single, GPU‑accelerated pipeline.
In the alignment stage, a hybrid algorithm combines global cross‑correlation with local fiducial‑based refinement, achieving angular errors below 0.5° and translational errors under 2 nm without manual intervention. Motion correction is performed on a per‑tilt basis using frame‑by‑frame alignment on direct‑electron detector movies, with GPU acceleration enabling near‑real‑time processing and preserving high‑frequency signal. The CTF estimation module treats each tilt angle independently, modeling angle‑dependent defocus and astigmatism by calculating the effective specimen thickness for each projection; this dynamic approach retains information up to 0.8 Å⁻¹.
Particle detection leverages a 3D ResNet‑based deep‑learning detector trained on a diverse set of purified complexes and cellular tomograms, reaching an average detection precision of ~92 %. Detected volumes are down‑sampled for rapid initial alignment, after which a multi‑scale refinement iteratively optimizes rotation and translation parameters.
The core of the workflow is the subtomogram alignment and averaging stage, which merges RELION’s Bayesian alignment framework with EMAN2’s multi‑class classification. Crucially, the authors feed CTF‑corrected complex‑valued volumes directly into the alignment, bypassing the traditional CTF back‑projection step that can introduce phase errors. Gold‑standard Fourier‑Shell Correlation (FSC) at the 0.143 criterion is used to assess resolution.
Benchmarking on two representative datasets demonstrates the system’s impact. For a 70 kDa purified marker complex, the automated pipeline improves the final resolution from 3.2 nm (manual processing) to 2.4 nm. For an in‑situ human ribosome, the workflow achieves 0.9 nm resolution, surpassing the 1.1 nm obtained with conventional methods, while reducing total processing time from roughly six days to under one day.
The authors discuss limitations, noting that while automatic parameter tuning works for most samples, highly heterogeneous or membrane‑rich specimens may still require manual fine‑tuning, and the heavy reliance on GPU resources necessitates access to high‑performance computing clusters. Future directions include real‑time streaming integration, multi‑scale particle classification plugins, and cloud‑based deployment to broaden accessibility.
In conclusion, this integrated workflow dramatically lowers the human effort required for cryo‑ET data processing, boosts throughput, and consistently delivers state‑of‑the‑art resolutions for both purified macromolecules and cellular contexts. By releasing the software as open source with standardized interfaces, the authors provide the structural biology community with a scalable platform that can be readily extended and adopted worldwide.
Comments & Academic Discussion
Loading comments...
Leave a Comment