FPGA Acceleration of Sequence Alignment: A Survey
Genomics is changing our understanding of humans, evolution, diseases, and medicines to name but a few. As sequencing technology is developed collecting DNA sequences takes less time thereby generating more genetic data every day. Today the rate of generating genetic data is outpacing the rate of computation power growth. Current sequencing machines can sequence 50 humans genome per day; however, aligning the read sequences against a reference genome and assembling the genome will take 1300 CPU hours. The main step in constructing the genome is aligning the reads against a reference genome. Numerous accelerators have been proposed to accelerate the DNA alignment process. Providing massive parallelism, FPGA-based accelerators have shown great performance in accelerating DNA alignment algorithms. Additionally, FPGA-based accelerators provide better energy efficiency than general-purpose processors. In this survey, we introduce three main DNA alignment algorithms and FPGA-based implementation of these algorithms to accelerate the DNA alignment. We also, compare these three alignment categories and show how accelerators are developing during the time.
💡 Research Summary
The paper “FPGA Acceleration of Sequence Alignment: A Survey” provides a comprehensive overview of how field‑programmable gate arrays (FPGAs) are being employed to speed up the most computationally intensive step in modern genomics pipelines: aligning short DNA reads to a reference genome. The authors begin by highlighting the dramatic imbalance between the rapid increase in sequencing throughput (e.g., a single Illumina HiSeq can generate data for up to 50 human genomes per day) and the comparatively slow growth of general‑purpose CPU performance. Aligning the resulting billions of reads can consume more than 1,300 CPU‑hours per run, making hardware acceleration a necessity.
The survey categorizes DNA alignment algorithms into three major families and examines representative FPGA implementations for each:
-
Pairwise Sequence Alignment (PSA) – This family includes classic dynamic‑programming algorithms such as Needleman‑Wunsch (global alignment) and Smith‑Waterman (local alignment). The authors explain that the alignment matrix H(i,j) is computed using a recurrence that depends on the three neighboring cells (up, left, diagonal). Because cells on the same diagonal are independent, FPGA designs exploit systolic arrays of Processing Elements (PEs) that compute an entire diagonal in parallel. Early work by Lipton et al. introduced a custom VLSI systolic array; later FPGA implementations refined the approach by simplifying scoring (e.g., +1 for match, –1 for mismatch, –2 for gaps), pipelining the data flow, and using on‑chip Block RAM to store scoring matrices. The paper discusses design challenges such as routing congestion for large PE arrays, memory bandwidth for streaming long reads, and the trade‑off between precision (full substitution matrices) and resource usage. Reported speed‑ups range from 10× to over 100× relative to optimized CPU code, with energy efficiency improvements of 5–20×.
-
Hash‑Table Based Alignment – Algorithms such as SSAHA and BLAST use k‑tuple hashing to locate exact or near‑exact matches between reads and the reference. The survey describes FPGA implementations that store massive hash tables in multi‑port BRAM or external DDR memory, generate k‑tuples on the fly, and perform parallel look‑ups. Collision resolution (chaining or open addressing) is realized in hardware, and the pipeline is designed to overlap hash generation, memory access, and candidate filtering. While this approach dramatically reduces the number of DP calculations, it incurs a high memory footprint for the hash structures. The authors note that several designs employ compressed hash representations (e.g., Bloom filters) to mitigate this issue. Performance gains of 5–30× over CPU baselines are reported, together with 3–10× lower power consumption.
-
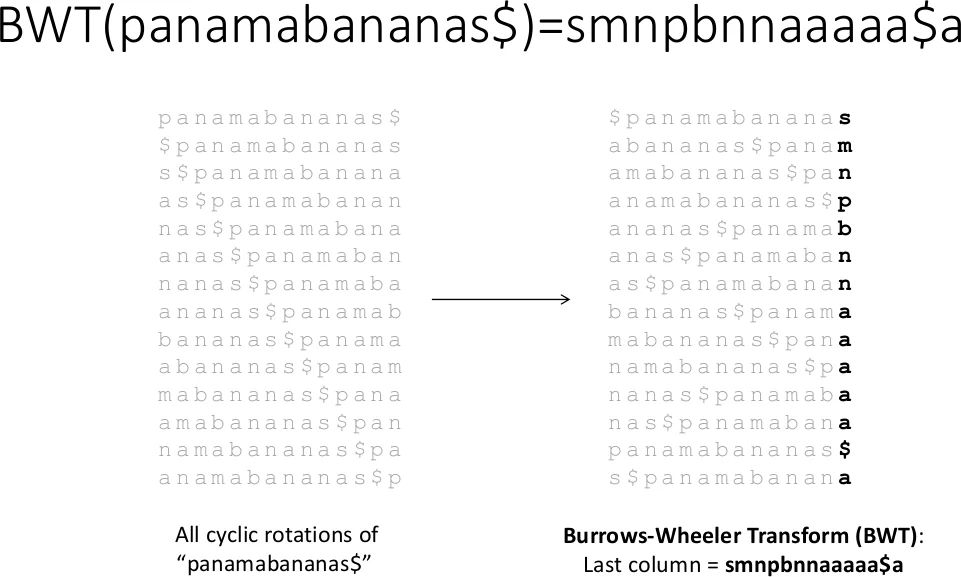
Burrows‑Wheeler Transform (BWT) with FM‑Index – Modern short‑read aligners (Bowtie, BWA, SOAP2) compress the reference genome using BWT and enable fast backward searches via the FM‑index. The survey details FPGA architectures that pre‑compute the i(s) and c(n,s) tables, store them in on‑chip memory, and implement the iterative “top/bottom” pointer update loop as a deeply pipelined unit. Because BWT reduces the reference size dramatically, memory bandwidth becomes the dominant bottleneck; the surveyed designs address this by using high‑speed interfaces (PCIe Gen3/4, HBM) and by parallelizing multiple query streams. Reported accelerations range from 10× to 50× for whole‑genome alignment, with memory traffic reduced to less than 30 % of the CPU requirement and energy efficiency gains of 8–25×.
The paper then provides a comparative table summarizing the strengths and weaknesses of each algorithmic family when mapped to FPGA hardware. Dynamic‑programming approaches deliver exact optimal alignments but demand large PE arrays and careful routing; hash‑based methods are fast for candidate generation but suffer from hash‑table size constraints; BWT‑based schemes excel in memory efficiency and scalability to large genomes but involve more complex control logic for the FM‑index traversal.
A timeline of key publications shows an evolution from early custom ASIC‑style systolic arrays to modern high‑level synthesis (HLS) designs and cloud‑based FPGA services. The authors also discuss emerging hybrid strategies, such as using BWT to prune the search space followed by a lightweight DP refinement on the FPGA, and multi‑FPGA clusters that share the workload via high‑bandwidth interconnects (e.g., CXL).
In conclusion, the survey confirms that FPGAs are uniquely suited to address the computational bottleneck of DNA read alignment. By tailoring the hardware architecture to the algorithmic characteristics—exploiting parallel diagonals, hash parallelism, or compressed index traversal—researchers can achieve orders‑of‑magnitude speed‑ups while dramatically lowering energy consumption. The paper calls for continued exploration of hybrid algorithms, better memory hierarchies, and integration with emerging sequencing technologies (e.g., long‑read platforms) to further close the gap between data generation and analysis.
Comments & Academic Discussion
Loading comments...
Leave a Comment