Formal Concept Analysis for Knowledge Discovery from Biological Data
Due to rapid advancement in high-throughput techniques, such as microarrays and next generation sequencing technologies, biological data are increasing exponentially. The current challenge in computational biology and bioinformatics research is how to analyze these huge raw biological data to extract biologically meaningful knowledge. This review paper presents the applications of formal concept analysis for the analysis and knowledge discovery from biological data, including gene expression discretization, gene co-expression mining, gene expression clustering, finding genes in gene regulatory networks, enzyme/protein classifications, binding site classifications, and so on. It also presents a list of FCA-based software tools applied in biological domain and covers the challenges faced so far.
💡 Research Summary
The reviewed paper provides a comprehensive overview of how Formal Concept Analysis (FCA), a lattice‑theoretic method for binary relational data, can be employed to extract biologically meaningful knowledge from the massive amounts of data generated by high‑throughput technologies such as microarrays and next‑generation sequencing. After outlining the exponential growth of biological databases (primary, secondary, and composite) and the challenges of handling genomics, transcriptomics, and proteomics data, the authors focus on several key application domains.
In transcriptomics, FCA is used to discretize continuous gene‑expression values (e.g., over‑expressed = 1, under‑expressed = 0) and to construct formal contexts via interval‑based, interordinal, or pattern‑structure scaling. Studies by Kaytoue‑Uberall and colleagues demonstrate that interval‑FCA and pattern‑structure FCA can efficiently identify co‑expressed gene groups even in large datasets (≈22 000 genes, 5 conditions). The paper also describes a workflow for DNA‑methylation analysis in breast‑cancer subtypes, where statistical filtering (normality tests, t‑test or Wilcoxon) precedes FCA lattice construction using the ConExp tool, revealing subtype‑specific hypomethylated gene patterns.
For clustering, FCA offers an alternative to hierarchical, k‑means, SOM, or fuzzy c‑means methods. Choi et al. build lattices for each experiment, define distance measures (including spectral and maximal common sub‑lattice distances), and compare lattices to assess similarity among experiments. Melo et al. integrate FCA with association‑rule mining and visual analytics in the CUBIST platform, enabling interactive exploration of large expression datasets.
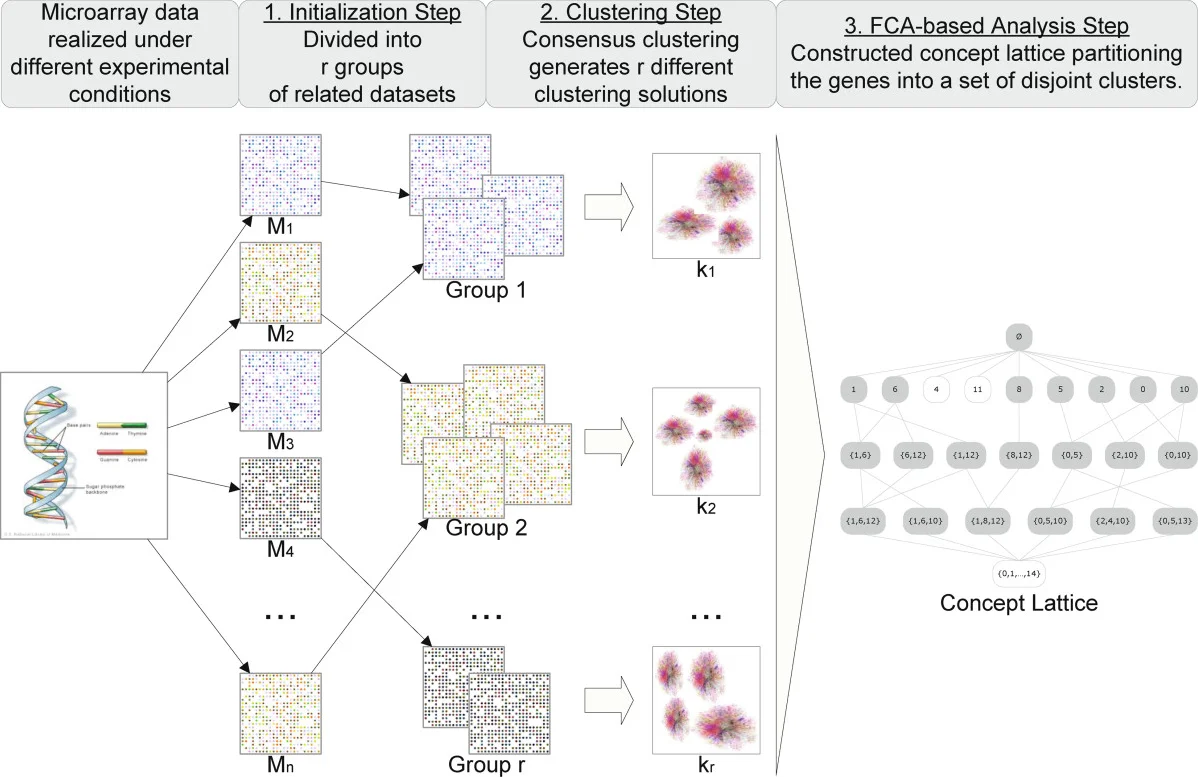
When multiple microarray experiments are available, FCA can consolidate consensus clustering results. Hristoskova’s FCA‑enhanced consensus clustering partitions genes across heterogeneous datasets, allowing each experiment group to retain its own gene set and avoiding the loss of information inherent in intersect‑only approaches. Benabderrahmane extends this idea to symbolic data mining, combining expression profiles with GO terms, pathway information, and interaction networks to perform biclustering and disease‑similarity analysis.
Disease similarity studies illustrate FCA’s ability to capture multi‑disease relationships algebraically. Keller et al. construct concepts from gene‑disease associations, producing lattices that reveal hidden connections among diseases sharing gene sets, and allowing the incorporation of additional annotations to refine these relationships.
In the context of gene regulatory networks (GRNs), Gebert and collaborators propose an FCA‑based pipeline that uses known interaction pairs and time‑series expression matrices to identify unknown network members. By treating interaction data as a binary relation and expression profiles as attributes, the method extracts candidate genes that strongly associate with a seed set, demonstrating FCA’s utility for dynamic network inference.
Overall, the paper highlights FCA’s strengths: its capacity to transform heterogeneous biological data into interpretable lattice structures, its compatibility with statistical preprocessing, and its potential to integrate diverse annotations (GO, KEGG, protein‑protein interactions). It also acknowledges practical limitations such as lattice explosion in high‑dimensional settings, the need for effective scaling strategies, and the filtering of non‑informative concepts. Future directions include algorithmic optimizations, hybrid approaches combining FCA with machine‑learning techniques, and the development of more user‑friendly visualization tools to make FCA accessible to a broader bioinformatics community.
Comments & Academic Discussion
Loading comments...
Leave a Comment