Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems
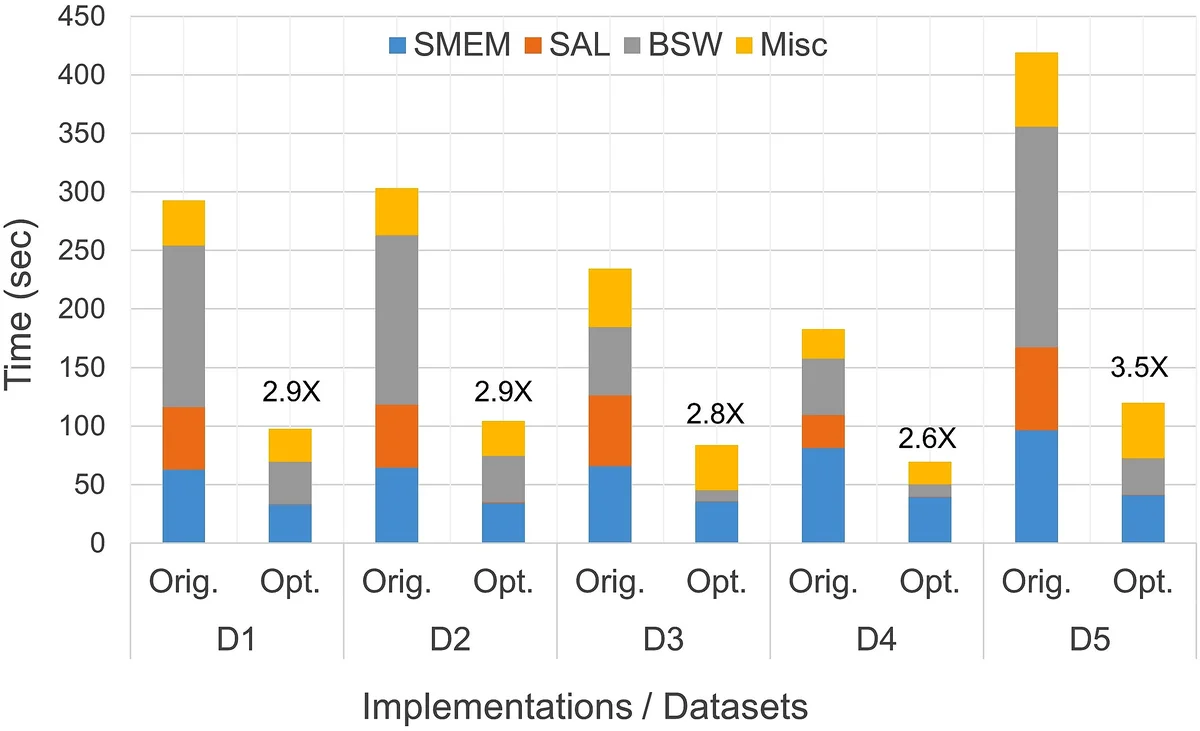
Innovations in Next-Generation Sequencing are enabling generation of DNA sequence data at ever faster rates and at very low cost. Large sequencing centers typically employ hundreds of such systems. Such high-throughput and low-cost generation of data underscores the need for commensurate acceleration in downstream computational analysis of the sequencing data. A fundamental step in downstream analysis is mapping of the reads to a long reference DNA sequence, such as a reference human genome. Sequence mapping is a compute-intensive step that accounts for more than 30% of the overall time of the GATK workflow. BWA-MEM is one of the most widely used tools for sequence mapping and has tens of thousands of users. In this work, we focus on accelerating BWA-MEM through an efficient architecture aware implementation, while maintaining identical output. The volume of data requires distributed computing environment, usually deploying multicore processors. Since the application can be easily parallelized for distributed memory systems, we focus on performance improvements on a single socket multicore processor. BWA-MEM run time is dominated by three kernels, collectively responsible for more than 85% of the overall compute time. We improved the performance of these kernels by 1) improving cache reuse, 2) simplifying the algorithms, 3) replacing small fragmented memory allocations with a few large contiguous ones, 4) software prefetching, and 5) SIMD utilization wherever applicable - and massive reorganization of the source code enabling these improvements. As a result, we achieved nearly 2x, 183x, and 8x speedups on the three kernels, respectively, resulting in up to 3.5x and 2.4x speedups on end-to-end compute time over the original BWA-MEM on single thread and single socket of Intel Xeon Skylake processor. To the best of our knowledge, this is the highest reported speedup over BWA-MEM.
💡 Research Summary
The paper addresses the growing computational bottleneck in next‑generation sequencing (NGS) pipelines, where mapping short reads to a reference genome consumes more than 30 % of the total runtime in the GATK best‑practice workflow. BWA‑MEM, a widely adopted read‑mapper, is the focus of this study. Although BWA‑MEM already balances speed and accuracy, its original implementation suffers from irregular memory accesses, heavy branching, and a lack of vectorization, limiting its performance on modern multicore CPUs.
Through meticulous profiling of BWA‑MEM version 0.7.25, the authors identify three kernels—Super‑maximal Exact Match search (SMEM), Suffix‑Array Lookup (SAL), and Banded Smith‑Waterman (BSW)—as responsible for over 85 % of the total execution time. Detailed analysis reveals that SMEM’s use of a heavily compressed FM‑index leads to ~285 k instructions per read and high LLC miss rates; SAL’s compressed suffix array forces ~5 k instructions per lookup; and BSW’s scalar implementation is instruction‑bound due to extensive branching and short loops.
To overcome these bottlenecks while preserving bit‑identical output, the authors apply a suite of architecture‑aware optimizations:
-
Workflow Reorganization – Instead of processing each read through all stages sequentially, reads are grouped into batches. Each stage (SMEM, SAL, BSW) processes an entire batch before moving to the next stage. This batch‑oriented design enables SIMD parallelism across reads and simplifies OpenMP dynamic scheduling.
-
Memory Allocation Refactoring – Frequent small allocations are replaced by a few large, contiguous memory pools that are reused across batches. This change improves hardware prefetching, reduces allocation overhead, and enhances cache reuse.
-
FM‑Index Tuning – The O‑bucket size η is chosen as a power of two, allowing division and modulo operations to be replaced with bit‑shifts and masks. Base symbols are encoded in 2‑bit form, and count queries are performed with bitwise operations, dramatically reducing instruction count in SMEM.
-
Algorithmic Simplification & Software Prefetch – Redundant loops and conditionals in SMEM are eliminated, and explicit software prefetch instructions are inserted to raise L1/L2 hit rates.
-
Suffix‑Array Decompression – The compressed suffix array is unpacked into a contiguous array, turning a costly indirect lookup into a simple indexed read, cutting per‑lookup instruction count from thousands to a few dozen.
-
SIMD Vectorization of BSW – The BSW dynamic‑programming core is rewritten using AVX‑512 intrinsics, processing multiple cells of the DP matrix in parallel. Loop unrolling and vector‑friendly data layout reduce branch mispredictions and achieve an 8× speedup.
Performance evaluation on an Intel Xeon Skylake single socket and an Intel Xeon E5 v3 platform demonstrates dramatic gains: SMEM runs 2× faster, SAL astonishingly 183× faster, and BSW 8× faster. Overall, the optimized BWA‑MEM2 delivers up to 3.5× speedup on a single thread and 2.4× speedup on a fully threaded single‑socket run, all while producing output identical to the original tool.
The authors emphasize that their optimizations are generic to modern multicore CPUs and do not rely on specialized hardware such as GPUs or FPGAs. Maintaining exact output is crucial for long‑running genomics studies, and this work shows that substantial performance improvements are achievable without sacrificing reproducibility. Future work is suggested in extending these techniques to multi‑socket systems, exploring dynamic load balancing across sockets, and integrating the optimized kernels into heterogeneous accelerator environments.
Comments & Academic Discussion
Loading comments...
Leave a Comment