Parallel Accelerated Custom Correlation Coefficient Calculations for Genomics Applications
The massive quantities of genomic data being made available through gene sequencing techniques are enabling breakthroughs in genomic science in many areas such as medical advances in the diagnosis and treatment of diseases. Analyzing this data, however, is a computational challenge insofar as the computational costs of the relevant algorithms can grow with quadratic, cubic or higher complexity-leading to the need for leadership scale computing. In this paper we describe a new approach to calculations of the Custom Correlation Coefficient (CCC) between Single Nucleotide Polymorphisms (SNPs) across a population, suitable for parallel systems equipped with graphics processing units (GPUs) or Intel Xeon Phi processors. We describe the mapping of the algorithms to accelerated processors, techniques used for eliminating redundant calculations due to symmetries, and strategies for efficient mapping of the calculations to many-node parallel systems. Results are presented demonstrating high per-node performance and near-ideal parallel scalability with rates of more than nine quadrillion elementwise comparisons achieved per second with the latest optimized code on the ORNL Titan system, this being orders of magnitude faster than rates achieved using other codes and platforms as reported in the literature. Also it is estimated that as many as 90 quadrillion comparisons per second may be achievable on the upcoming ORNL Summit system, an additional 10X performance increase. In a companion paper we describe corresponding techniques applied to calculations of the Proportional Similarity metric for comparative genomics applications.
💡 Research Summary
The paper addresses the computational bottleneck inherent in calculating the Custom Correlation Coefficient (CCC) for large‑scale genomic data sets. CCC quantifies the co‑occurrence of alleles across pairs (2‑way) or triples (3‑way) of single‑nucleotide polymorphisms (SNPs), taking into account partial population structures and genetic heterogeneity. While biologically powerful, the naïve computation scales as O(n_f n_v²) for 2‑way and O(n_f n_v³) for 3‑way, where n_f is the number of samples and n_v the number of SNPs, quickly exceeding the capacity of conventional clusters.
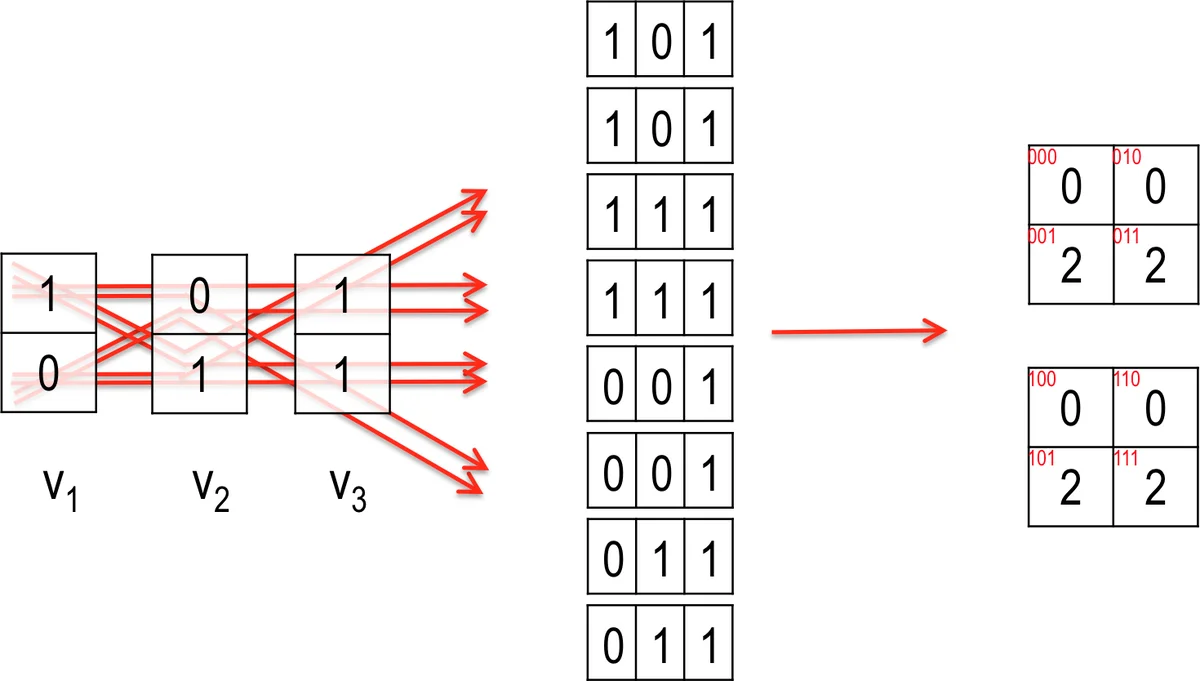
The authors propose a suite of algorithms that map these calculations onto modern many‑core accelerators—NVIDIA GPUs and Intel Xeon Phi coprocessors—by recasting the core tallying operation as a modified dense matrix‑matrix multiplication (GEMM). For the 2‑way case, each SNP entry (a 2‑bit allele pair) is packed into 64‑bit words, allowing 64 SNPs per word. Bit‑wise shifts, logical AND/OR, and the hardware population‑count (popcnt) instruction are then used to simultaneously update the four entries of a 2 × 2 allele‑pair frequency table. The resulting kernel, called mGEMM2, reuses the highly tuned ZGEMM routine from the MAGMA library, storing the four integer tallies in the mantissas of two double‑precision numbers (25 bits each). This design avoids overflow for up to 8 388 607 samples, far beyond typical study sizes.
The 3‑way CCC cannot be expressed directly as a single GEMM because the required tally table would exceed the mantissa capacity. The authors therefore introduce a two‑step method. First, for a chosen reference SNP v_j, three auxiliary matrices X_{j,1}, X_{j,2}, X_{j,3} are constructed by recoding the allele pairs according to a lookup table (Table 1). Each X matrix encodes a different conditional mapping of v_j against the full SNP matrix V. Second, a modified GEMM (mGEMM3) is performed between each X_{j,ξ}ᵀ and V, yielding intermediate 2‑way tallies that can be algebraically combined to reconstruct the full 3‑way frequency counts. The same bit‑packing and popcnt strategy is employed, preserving the high arithmetic intensity of the operation.
Parallelism across nodes is achieved by partitioning the SNP set into blocks and assigning each block pair (or triple) to a distinct MPI rank. Symmetry (i < j < k) eliminates redundant work, and asynchronous MPI communication overlaps data exchange with computation, delivering near‑ideal strong scaling. On the ORNL Titan system (18 688 nodes, each with a K80 GPU), the implementation reaches 3.2 × 10¹¹ pairwise comparisons per node per second, aggregating to more than 9 × 10¹⁵ element‑wise comparisons per second for the 2‑way CCC. Scaling tests up to 1 024 nodes retain >95 % parallel efficiency, outperforming prior GPU‑only codes (e.g., GBOOST, GWISFI) by an order of magnitude or more. Memory usage is minimized by storing SNPs in a 2‑bit format, enabling billions of SNPs to reside within a single node’s memory.
Performance projections for the upcoming Summit supercomputer (IBM Power9 CPUs + NVIDIA V100 GPUs) suggest a tenfold increase in throughput, potentially achieving 9 × 10¹⁶ comparisons per second. The paper concludes that the combination of (i) algorithmic reformulation as modified GEMM kernels, (ii) aggressive bit‑level packing with hardware popcnt, (iii) symmetry‑driven redundancy elimination, and (iv) scalable MPI orchestration constitutes a breakthrough for large‑scale genomic correlation analysis. Future work will explore extensions to higher‑order (k‑way) CCC, further memory‑bandwidth optimizations, and deployment on cloud‑based heterogeneous clusters.
Comments & Academic Discussion
Loading comments...
Leave a Comment