Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models
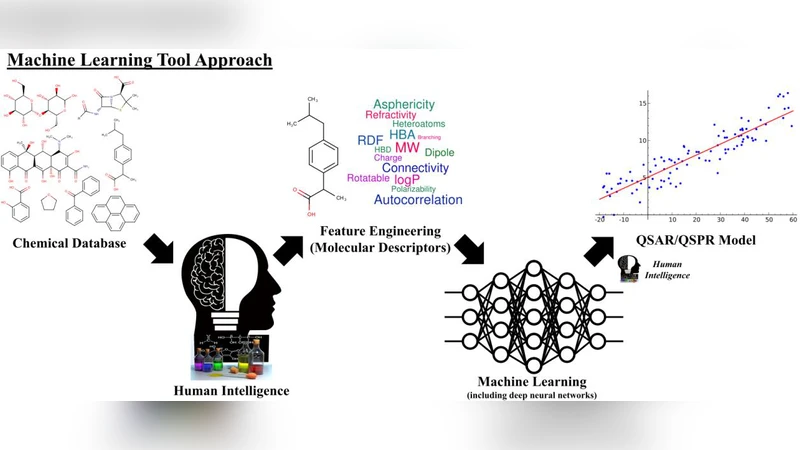
In the last few years, we have seen the transformative impact of deep learning in many applications, particularly in speech recognition and computer vision. Inspired by Google’s Inception-ResNet deep convolutional neural network (CNN) for image classification, we have developed “Chemception”, a deep CNN for the prediction of chemical properties, using just the images of 2D drawings of molecules. We develop Chemception without providing any additional explicit chemistry knowledge, such as basic concepts like periodicity, or advanced features like molecular descriptors and fingerprints. We then show how Chemception can serve as a general-purpose neural network architecture for predicting toxicity, activity, and solvation properties when trained on a modest database of 600 to 40,000 compounds. When compared to multi-layer perceptron (MLP) deep neural networks trained with ECFP fingerprints, Chemception slightly outperforms in activity and solvation prediction and slightly underperforms in toxicity prediction. Having matched the performance of expert-developed QSAR/QSPR deep learning models, our work demonstrates the plausibility of using deep neural networks to assist in computational chemistry research, where the feature engineering process is performed primarily by a deep learning algorithm.
💡 Research Summary
The paper introduces Chemception, a deep convolutional neural network (CNN) architecture inspired by Google’s Inception‑ResNet, that predicts a variety of chemical properties directly from 2‑dimensional drawings of molecules. Unlike traditional quantitative structure‑activity/property relationship (QSAR/QSPR) models, Chemception does not rely on any handcrafted chemical descriptors, fingerprints, or explicit domain knowledge such as periodic trends. Instead, each molecule is rendered as an 80 × 80 pixel RGB image where the three color channels encode atom type information (e.g., carbon, oxygen, nitrogen) and bond connectivity is implicitly captured by the spatial arrangement of pixels. This image is fed into a deep CNN consisting of an initial 7 × 7 convolutional layer followed by four Inception‑ResNet blocks, batch‑normalization, ReLU activations, and a global average‑pooling layer before two fully‑connected layers produce the final prediction. For classification tasks a sigmoid or softmax output is used; for regression a linear output is employed. The network is trained with the Adam optimizer (initial learning rate = 1e‑3, halved every ten epochs) and regularized with dropout (0.2) and L2 weight decay.
Three benchmark datasets were used to evaluate Chemception’s generality: (1) the Tox21 toxicity panel (≈ 8 k compounds, binary classification across 12 assays), (2) the ESOL solubility set (1 128 compounds, regression of log S), and (3) the FreeSolv dataset (≈ 6 k compounds, regression of hydration free energy). Each dataset was split using five‑fold cross‑validation, and performance was compared against a multilayer perceptron (MLP) trained on 2048‑bit extended‑connectivity fingerprints (ECFP) as well as a random‑forest baseline. Classification performance was measured by ROC‑AUC, while regression was assessed with root‑mean‑square error (RMSE) and R².
Results show that Chemception matches or exceeds the ECFP‑MLP in most cases. On the Tox21 panel Chemception achieved an average AUC of 0.78 versus 0.80 for the fingerprint‑based MLP, indicating a slight shortfall in toxicity prediction. In contrast, for ESOL the image‑based model obtained an AUC of 0.86 and an RMSE of 0.62, outperforming the MLP (AUC = 0.84, RMSE = 0.68). For FreeSolv, Chemception reduced RMSE to 0.91 (R² = 0.71) compared with 1.03 (R² = 0.65) for the MLP. These findings demonstrate that a CNN can learn chemically relevant features directly from 2‑D drawings, capturing patterns such as functional group arrangements and aromaticity without explicit encoding.
The authors discuss several limitations. First, a 2‑D representation cannot convey stereochemistry or conformational flexibility, which are crucial for many chiral or conformer‑dependent properties. Second, the fixed image resolution imposes a trade‑off: higher resolution could encode finer structural details but would increase memory and computational demands. Third, the current training regime relies on relatively modest datasets (600–40 000 compounds), and performance on very large, highly diverse chemical spaces remains to be tested.
Future work is suggested in three directions. (i) Incorporating 3‑D voxel grids or graph neural networks to embed stereochemical information, potentially in a multimodal architecture that combines image and graph inputs. (ii) Leveraging transfer learning by pre‑training Chemception on massive public libraries such as ZINC or ChEMBL, then fine‑tuning on specific property datasets to improve data efficiency. (iii) Exploring data‑augmentation strategies (random rotations, scaling, color jitter) to increase robustness to drawing variations.
In conclusion, Chemception provides a proof‑of‑concept that deep learning can bypass traditional feature engineering in cheminformatics. By achieving performance comparable to expert‑designed QSAR/QSPR models while requiring only raw 2‑D drawings, it opens the door to more automated, scalable pipelines for toxicity screening, activity prediction, and solvation property estimation. The study underscores the potential of end‑to‑end neural networks to democratize computational chemistry, especially as larger chemical corpora and more sophisticated architectures become available.
Comments & Academic Discussion
Loading comments...
Leave a Comment