Observations and perspectives on the diversification of genomes
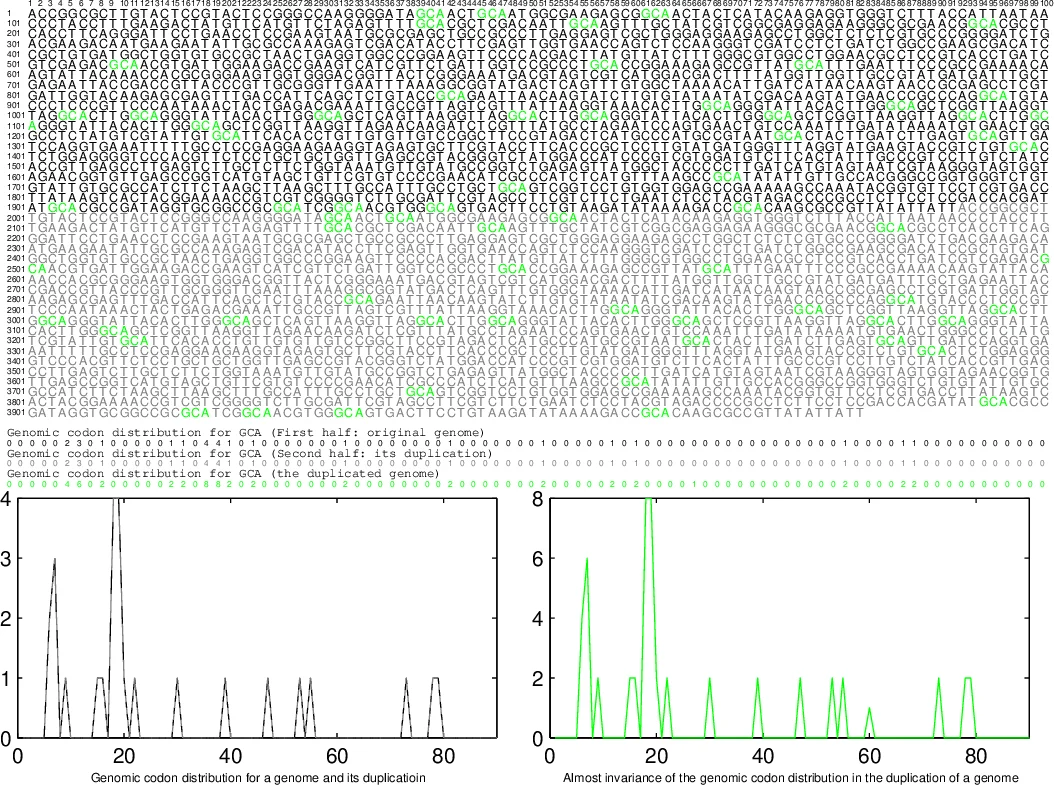
Rich information on the prebiotic evolution is still stored in contemporary genomic data. The statistical mechanism at the sequence level may play a significant role in the prebiotic evolution. Based on statistical analysis of genome sequences, it has been observed that there is a close relationship between the evolution of the genetic code and the organisation of genomes. A biodiversity space for species is constructed based on comparing the distributions of codons in genomes for different species according to recruitment order of codons in the prebiotic evolution, by which a closely relationship between the evolution of the genetic code and the tree of life has been confirmed. On one hand, the three domain tree of life can be reconstructed according to the distance matrix of species in this biodiversity space, which supports the three-domain tree rather than the eocyte tree. On the other hand, an evolutionary tree of codons can be obtained by comparing the distributions of the 64 codons in genomes, which agrees with the recruitment order of codons on the roadmap. This is a simple phylogenomic method to study the origins of metazoan, the evolution of primates, etc. This study should be regarded as an exploratory attempt to explain the diversification of the three domains of life by statistical mechanism in prebiotic sequence evolution. It is indicated that the number of bases in the triplet codons might be explained statistically by the number of strands in the triplex DNAs. The adaptation of life to the changing environment might be due to assembly of redundant genomes at the sequence level.
💡 Research Summary
The paper presents an exploratory framework that links modern genomic codon usage patterns to a hypothesized pre‑biotic “codon recruitment roadmap” and to the large‑scale diversification of life into the three domains (Bacteria, Archaea, Eukarya). The authors begin by defining a “genomic codon distribution” for each fully sequenced organism: a normalized set of 64 frequency profiles describing the inter‑codon distances across the entire genome. They argue that these distributions reveal a universal genome format shared by all life, including viruses.
Using a previously proposed order in which the 64 codons were recruited during early molecular evolution, the authors construct three quantitative descriptors for each species: route bias (which recruitment pathway a codon belongs to), hierarchy bias (its position in the hierarchical recruitment ladder), and fluctuation amplitude (the magnitude of three‑base periodic fluctuations in the codon distance profile). Plotting each organism in a three‑dimensional Euclidean space defined by these descriptors yields a “biodiversity space.” Pairwise Euclidean distances in this space generate a distance matrix that, when subjected to standard phylogenetic clustering, reproduces a tree of life that matches the classic three‑domain topology. Notably, the resulting tree places Euryarchaeota closer to Crenarchaeota than to Eukaryotes, thereby supporting the three‑domain model over the eocyte hypothesis.
In parallel, the authors treat the 64 codons themselves as “taxa” and cluster them based on the average codon‑distribution vectors across all genomes. The resulting codon phylogeny mirrors the recruitment order proposed in the earlier “roadmap” papers (Li 2018‑I), suggesting that modern codon usage retains a fossil record of early codon addition events.
A central, highly speculative component of the work is the “triplex‑to‑duplex” picture. The authors posit that early nucleic acids existed as triple‑helix (triplex) structures. Because a single‑stranded DNA derived from a triplex would be three times longer, they argue that the number three became the natural divisor for defining codon length, explaining why the genetic code is triplet‑based. They further claim that the three‑base periodic fluctuations observed in codon distributions arise from random assembly of incomplete codon subsets during successive stages of the recruitment roadmap. The paper cites structural analyses of early proteins (modules of ~25–30 amino acids) as supporting evidence for a stepwise assembly of longer genes from short motifs encoded by triplex‑derived sequences.
The authors also introduce three “bias” concepts—route bias, hierarchy bias, and fluctuation amplitude—to quantify differences among species, domains, and viruses. They argue that viruses represent the earliest life forms because their genomes could be assembled from the smallest, earliest codon subsets. The separation of Bacteria and Archaea is attributed to “leaf‑node bias,” while the divergence of Archaea and Eukarya is linked to fluctuation amplitude. The transition from unicellular to multicellular organisms is suggested to have occurred when eukaryotic genomes began to incorporate quasi‑complete codon subsets.
Methodologically, the study relies heavily on the computation of normalized codon‑distance vectors, Euclidean distance calculations, and clustering algorithms (though the specific algorithms and parameter settings are not detailed). The data source is the complete genome collection in GenBank, and taxonomic classification follows NCBI taxonomy. The authors claim that their approach provides a “general phylogenomic method” applicable to questions such as metazoan origins and primate evolution, but no concrete case studies or statistical validation are presented.
Critical assessment reveals several weaknesses. First, the recruitment order of codons is taken as a given hypothesis from earlier papers, without independent biochemical or experimental support. Second, the statistical pipeline lacks transparency: the normalization of codon distance distributions, handling of genome size bias, and choice of clustering method are insufficiently described, limiting reproducibility. Third, the triplex‑DNA hypothesis, while imaginative, is not substantiated by experimental evidence of stable triplex structures under plausible pre‑biotic conditions, nor does it explain why a three‑strand system would be favored over other oligomeric forms. Fourth, the claim that the three‑domain tree is uniquely recovered from codon‑distribution data is not contrasted with alternative trees generated from the same data using different distance metrics or weighting schemes; thus, potential data‑selection bias remains unaddressed.
In summary, the paper offers a novel perspective by treating codon usage patterns as a fossil record of early molecular evolution and by constructing a multidimensional “biodiversity space” that recapitulates the three‑domain phylogeny. However, the work is largely hypothesis‑driven, with limited empirical validation, opaque statistical methodology, and speculative mechanistic explanations (triplex‑to‑duplex transition). Future research should aim to (i) experimentally test the stability and formation pathways of triplex nucleic acids under pre‑biotic conditions, (ii) provide a fully reproducible computational pipeline with benchmark comparisons to alternative phylogenetic reconstructions, and (iii) assess the robustness of the biodiversity‑space approach across diverse genomic datasets and evolutionary timescales.
Comments & Academic Discussion
Loading comments...
Leave a Comment