QuickProbs 2: towards rapid construction of high-quality alignments of large protein families
Increasing size of sequence databases caused by the development of high throughput sequencing, poses multiple alignment algorithms to face one of the greatest challenges yet. As we show, well-established techniques employed for increasing alignment quality, i.e., refinement and consistency, are ineffective when large protein families are of interest. We present QuickProbs 2, an algorithm for multiple sequence alignment. Based on probabilistic models, equipped with novel column-oriented refinement and selective consistency, it offers outstanding accuracy. When analysing hundreds of sequences, QuickProbs 2 is significantly better than Clustal Omega, the previous leader for processing numerous protein families. In the case of smaller sets, for which consistency-based methods are the best performing, QuickProbs 2 is also superior to the competitors. Due to computational scalability of selective consistency and utilisation of massively parallel architectures, presented algorithm is comparable to Clustal Omega in terms of execution time, and orders of magnitude faster than full consistency approaches, like MSAProbs or PicXAA. All these make QuickProbs 2 a useful tool for aligning families ranging from few, to hundreds of proteins. QuickProbs 2 is available at https://github.com/refresh-bio/QuickProbs.
💡 Research Summary
The paper introduces QuickProbs 2, a novel multiple sequence alignment (MSA) algorithm designed to handle the explosive growth of protein sequence databases generated by high‑throughput sequencing technologies. While traditional MSA pipelines rely on two well‑established techniques—refinement (iterative correction of a progressive alignment) and consistency (incorporating information from all pairwise alignments into each alignment step)—the authors demonstrate that both become ineffective or even detrimental when the number of sequences exceeds a few hundred.
QuickProbs 2 builds on its predecessor QuickProbs, which already leveraged probabilistic models (pair‑hidden Markov models and partition functions) and GPU acceleration to achieve speed comparable to Clustal Ω while retaining the accuracy of consistency‑based methods. The new version introduces two key algorithmic innovations:
-
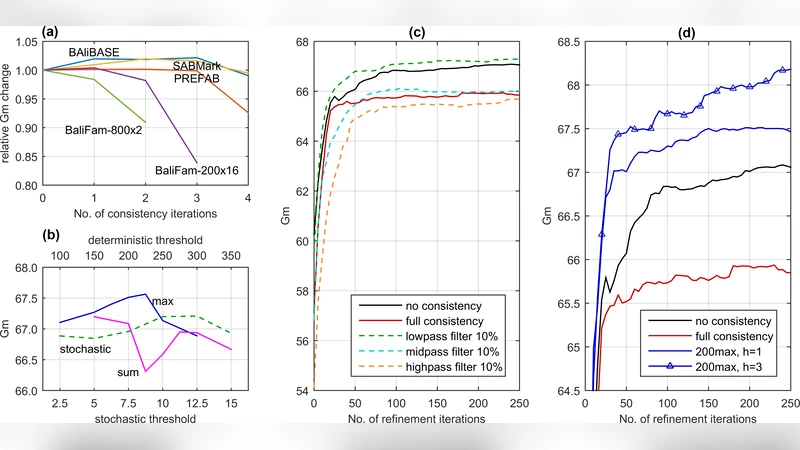
Column‑oriented refinement – Instead of splitting the alignment randomly or by cutting a random branch in the guide tree (as done in earlier tools such as MAFFT or MUSCLE), QuickProbs 2 selects a column that contains at least one gap and divides the alignment into two profiles based on the presence or absence of a gap in that column. Because at least one profile is guaranteed to shrink, the chance of generating a different, higher‑quality alignment after realignment is dramatically increased. An additional acceptance criterion—non‑increasing alignment length—is used to drive convergence, based on the observation that erroneous alignments tend to be longer than correct ones.
-
Selective consistency – Full consistency requires O(k³n³) operations (k = number of sequences, n = average length) and quickly becomes dominated by noise for large families. QuickProbs 2 therefore relaxes each posterior probability matrix S_xy only through a subset of auxiliary sequences z that satisfy a distance‑based filter f(d_xz, d_yz) ≤ T. Two distance measures are explored: (a) a score‑based distance derived from the initial pairwise alignment scores, and (b) a tree‑guided distance equal to the number of nodes in the minimal subtree containing both x and y. Experiments show that tree‑guided filtering yields the best trade‑off between information gain and computational cost. The algorithm also rescales the signal of S_xy by a factor h_xy that depends on how many relaxations were performed, preventing over‑dilution of the original matrix in very large families.
Implementation-wise, QuickProbs 2 uses OpenCL to run on both NVIDIA and AMD GPUs, and can fall back to CPU execution when no accelerator is available. Posterior matrices are stored in a sparse format with a user‑adjustable sparsity coefficient β, allowing the program to fit within a user‑specified memory budget even for families containing up to 100 000 sequences. Additional improvements include replacing the Gonnet160 matrix with VTML200 for the partition‑function step, training gap penalties and temperature on the BALIBASE 3 benchmark using the NOMAD optimizer, and providing a nucleotide mode (HOXD substitution matrix and GTR evolutionary model).
The authors evaluated QuickProbs 2 on six benchmark collections: BALIBASE, PREFAB, extended OXBench, SABmark (all containing small to moderate families), and the large‑scale HomFam and BaliFam datasets (up to ~1 000 and ~100 000 sequences, respectively). Results show that:
- For small families (≤50 sequences) QuickProbs 2 outperforms all tested consistency‑based methods (MSAProbs, ProbCons, etc.) in both SP and TC scores.
- For medium‑size families (100–1 000 sequences) it consistently surpasses Clustal Ω, which is the current leader for large‑scale alignments.
- On the BaliFam benchmark (≈1 000 sequences) selective consistency adds a modest 2–3 % improvement over the non‑selective version.
- Execution time on a modern GPU is comparable to Clustal Ω and orders of magnitude faster than full‑consistency tools such as MSAProbs or PicXAA, even when the selective consistency step is enabled.
In summary, QuickProbs 2 delivers a rare combination of high alignment quality across all family sizes, scalable computational performance, and flexible resource usage. By redesigning refinement to be column‑oriented and by applying consistency only where it is most informative, the method overcomes the traditional trade‑off between accuracy and speed that has limited previous MSA tools. The open‑source implementation (https://github.com/refresh-bio/QuickProbs) makes it readily available for the bioinformatics community, and its ability to run on CPUs, GPUs, and in bulk‑processing mode further broadens its applicability to modern large‑scale proteomics and genomics projects.
Comments & Academic Discussion
Loading comments...
Leave a Comment