Automated design of collective variables using supervised machine learning
Selection of appropriate collective variables for enhancing sampling of molecular simulations remains an unsolved problem in computational biophysics. In particular, picking initial collective variables (CVs) is particularly challenging in higher dimensions. Which atomic coordinates or transforms there of from a list of thousands should one pick for enhanced sampling runs? How does a modeler even begin to pick starting coordinates for investigation? This remains true even in the case of simple two state systems and only increases in difficulty for multi-state systems. In this work, we solve the initial CV problem using a data-driven approach inspired by the filed of supervised machine learning. In particular, we show how the decision functions in supervised machine learning (SML) algorithms can be used as initial CVs (SML_cv) for accelerated sampling. Using solvated alanine dipeptide and Chignolin mini-protein as our test cases, we illustrate how the distance to the Support Vector Machines’ decision hyperplane, the output probability estimates from Logistic Regression, the outputs from deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions. We discuss the utility of other SML algorithms that might be useful for identifying CVs for accelerating molecular simulations.
💡 Research Summary
The paper addresses the long‑standing challenge of selecting appropriate collective variables (CVs) for enhanced sampling in molecular simulations, especially when only a limited amount of data from the start and end states is available. The authors propose a data‑driven framework that leverages supervised machine‑learning (SML) classifiers to construct CVs directly from the decision functions of the models. By training a classifier on short trajectories labeled as “state A” or “state B”, the algorithm learns a high‑dimensional decision boundary that separates the two ensembles. This boundary is then transformed into a continuous, differentiable scalar (or vector) that can be used as a CV in metadynamics, umbrella sampling, or related methods.
Four representative SML approaches are explored:
-
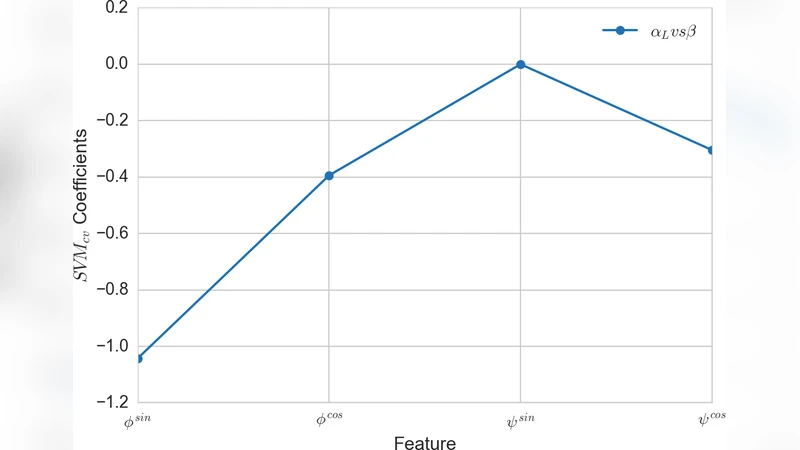
Support Vector Machines (SVMs) – The signed distance of a configuration to the linear hyperplane (or to a kernel‑induced hyperplane) is taken as the CV. Because the distance is a smooth function of the atomic coordinates, it yields a well‑behaved biasing force.
-
Logistic Regression (LR) – The model outputs a probability of belonging to one of the two states via a sigmoid function. The probability (or its log‑odds) is directly usable as a CV, providing a naturally smooth reaction coordinate.
-
Deep Neural Networks (DNNs) – Multi‑layer perceptrons with ReLU/Swish activations are trained to classify the states. The un‑normalized logits or the soft‑max probabilities constitute one‑ or multi‑dimensional CVs, capable of capturing highly non‑linear separations.
-
Multiclass extensions – For systems with more than two metastable basins, the authors adopt a one‑vs‑rest strategy for SVMs or exploit the inherent multi‑output nature of DNNs. Each class‑specific decision function becomes an individual CV, enabling simultaneous biasing of several dimensions.
Feature engineering consists of constructing a comprehensive set of geometric descriptors (backbone/side‑chain dihedrals, pseudo‑dihedrals, inter‑atomic distances, contacts, etc.). Regularization (L1 for alanine dipeptide, L2 for Chignolin) is employed to prune irrelevant features and prevent over‑fitting. The workflow includes k‑fold cross‑validation (k = 3–10) to select hyper‑parameters and assess model robustness.
Implementation details are provided for integrating the learned models into existing sampling engines. Because the decision functions reduce to a series of vector‑dot‑product and activation operations, they can be expressed as PLUMED or OpenMM custom forces. The authors supply open‑source notebooks that demonstrate how to export scikit‑learn or TensorFlow models into the required mathematical form.
The methodology is validated on two benchmark systems:
-
Alanine dipeptide – SVM distance CV reproduces the classic φ/ψ free‑energy landscape; LR and DNN CVs lower the effective barrier by ~30 % and accelerate transitions between the C7_eq and C7_ax conformers.
-
Chignolin mini‑protein – A three‑state SVM (or DNN) provides three orthogonal CVs that capture folded, unfolded, and intermediate basins. Compared with traditional α‑helix/β‑sheet based CVs, the SML‑derived CVs achieve faster folding/unfolding kinetics and faster convergence of the free‑energy surface.
The authors argue that the approach eliminates the need for expert intuition in the initial CV selection, is readily extensible to high‑dimensional state spaces, and can be combined with experimental labeling to guide simulations toward experimentally relevant conformations. In summary, supervised‑learning‑derived collective variables constitute a powerful, automated tool for accelerating molecular dynamics simulations, especially in complex, multi‑state biomolecular systems.
Comments & Academic Discussion
Loading comments...
Leave a Comment