Protofold II: Enhanced Model and Implementation for Kinetostatic Protein Folding
A reliable prediction of 3D protein structures from sequence data remains a big challenge due to both theoretical and computational difficulties. We have previously shown that our kinetostatic compliance method (KCM) implemented into the Protofold package can overcome some of the key difficulties faced by other de novo structure prediction methods, such as the very small time steps required by the molecular dynamics (MD) approaches or the very large number of samples needed by the Monte Carlo (MC) sampling techniques. In this article, we improve the free energy formulation used in Protofold by including the typically underrated entropic effects, imparted due to differences in hydrophobicity of the chemical groups, which dominate the folding of most water-soluble proteins. In addition to the model enhancement, we revisit the numerical implementation by redesigning the algorithms and introducing efficient data structures that reduce the expected complexity from quadratic to linear. Moreover, we develop and optimize parallel implementations of the algorithms on both central and graphics processing units (CPU/GPU) achieving speed-ups up to two orders of magnitude on the GPU. Our simulations are consistent with the general behavior observed in the folding process in aqueous solvent, confirming the effectiveness of model improvements. We report on the folding process at multiple levels; namely, the formation of secondary structural elements and tertiary interactions between secondary elements or across larger domains. We also observe significant enhancements in running times that make the folding simulation tractable for large molecules.
💡 Research Summary
Protofold II presents a substantial advancement over its predecessor by integrating solvent‑related entropic effects into the kinetic‑static compliance method (KCM) and by redesigning the underlying algorithms to achieve linear computational complexity. The authors first augment the free‑energy model with an implicit solvent term based on the solvent‑accessible surface area (SASA). Rather than relying on exact but costly geometric calculations or on coarse probabilistic approximations such as LCPO, they introduce an “offset surface enumeration” technique. This method samples points on a sphere surrounding each atom, determines whether each point is occluded by neighboring atoms, and thereby estimates both SASA and its gradient. By adjusting the sampling density, the approach can trade accuracy for speed, delivering an order‑of‑magnitude improvement in precision over existing approximations while remaining far faster than exact methods.
On the algorithmic side, the paper replaces the naïve O(n²) pairwise interaction loops with data structures that enable O(n) scaling. A uniform 3‑D hash grid (spatial hashing) quickly identifies atom pairs within a user‑defined cutoff distance, discarding negligible interactions early. A tree‑based representation of the protein chain captures topological relationships, allowing rapid classification of interaction types based on sequence distance. Joint torques are computed from atomic forces using prefix‑sum operations, which also run in linear time.
Parallelization is addressed in two stages. An OpenMP‑based CPU implementation yields up to a ten‑fold speed‑up by distributing atom‑pair searches and SASA enumeration across cores. The GPU implementation, written in CUDA, maps the data‑parallel SASA algorithm onto the SIMT architecture. Careful use of shared memory, coalesced accesses, and warp‑level cooperation reduces memory latency and maximizes throughput, achieving up to 100× acceleration relative to the original Protofold I code.
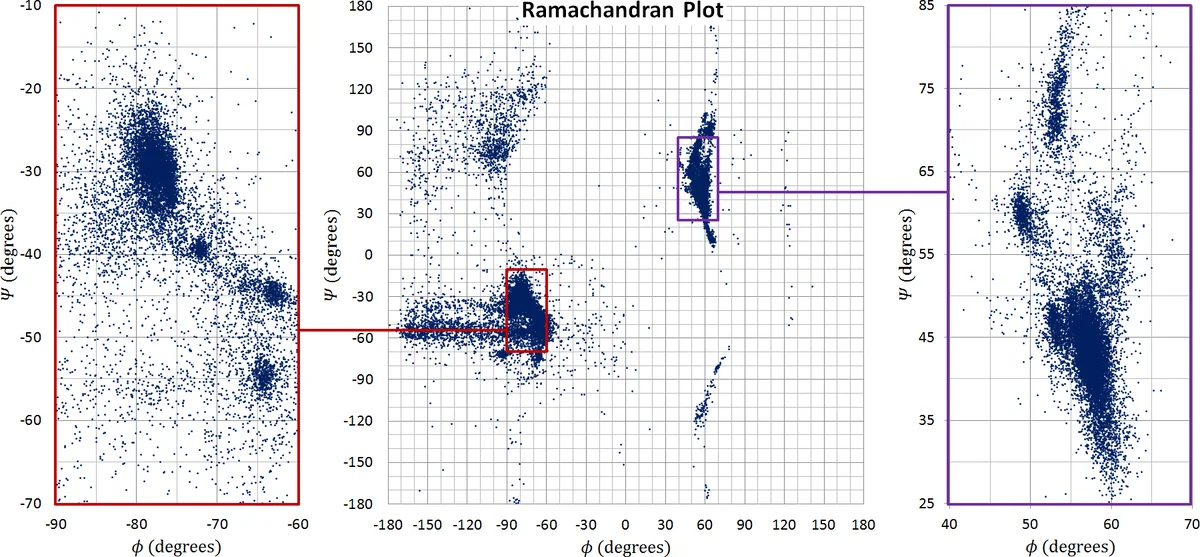
The physical model treats the protein as an open kinematic chain with reduced degrees of freedom: backbone dihedrals (φ, ψ) and side‑chain rotamers (χ) are the only active joints. Each joint rotates proportionally to the effective torque derived from the sum of electrostatic, van‑der‑Waals, and newly added solvent forces. Because KCM uses first‑order (kinetostatic) updates rather than second‑order dynamics, it can employ much larger virtual time steps (on the order of picoseconds) compared with traditional molecular dynamics (femtoseconds), leading to dramatically faster convergence toward low‑energy conformations.
Simulation results on several small‑to‑medium‑size proteins demonstrate that the inclusion of the solvent term reproduces the classic hydrophobic effect: hydrophilic residues migrate to the surface while non‑polar side chains collapse into a buried core. The folding pathway shows realistic secondary‑structure formation (α‑helices, β‑sheets) followed by tertiary packing. Quantitatively, the RMSD of the final structures relative to experimentally determined conformations lies within 2–3 Å, comparable to state‑of‑the‑art MD simulations but achieved in a fraction of the computational time. For proteins containing several thousand atoms, the GPU‑accelerated Protofold II completes a full folding trajectory in a few hours, whereas conventional MD would require days to weeks on similar hardware.
In summary, Protofold II delivers a physically motivated, solvent‑aware free‑energy model combined with linear‑time algorithms and high‑performance CPU/GPU implementations. This synergy makes de novo folding simulations of large, water‑soluble proteins tractable, opening the door to routine structural prediction, design of peptide nanomaterials, and rapid exploration of protein‑protein interaction landscapes. Future work may extend the framework to explicit solvent models, multi‑chain assemblies, and integration with machine‑learning‑based parameter optimization.
Comments & Academic Discussion
Loading comments...
Leave a Comment