Homochiral oligopeptides by chiral amplification: Interpretation of experimental data with a copolymerization model
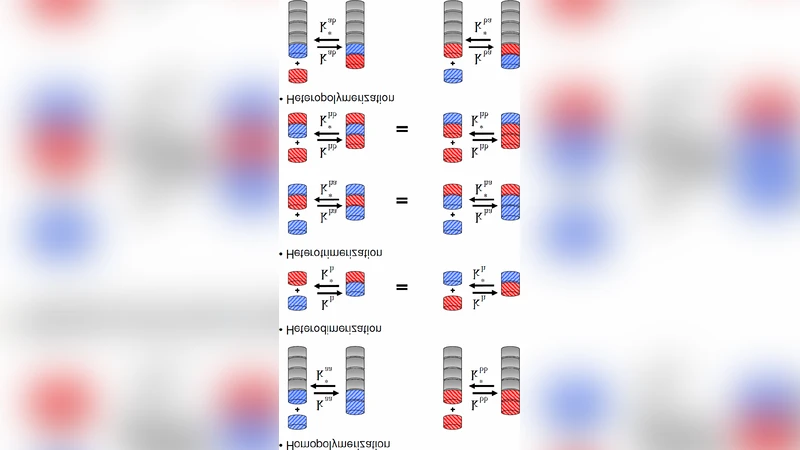
We present a differential rate equation model of chiral polymerization based on a simple copolymerization scheme in which the enantiomers are added to, or removed from, the homochiral or heterochiral chains (reversible stepwise isodesmic growth or dissociation). The model is set up for closed systems and takes into account the corresponding thermodynamic constraints implied by the reversible monomer attachments, while obeying a constant mass constraint. In its simplest form, the model depends on a single variable rate constant, the maximum chain length N, and the initial concentrations. We have fit the model to the experimental data from the Rehovot group on lattice-controlled chiral amplification of oligopeptides. We find in all the chemical systems employed except for one, that the model fits the measured relative abundances of the oligopetides with higher degrees of correlation than from a purely random polymerization process.
💡 Research Summary
The paper addresses the longstanding problem of how small initial enantiomeric imbalances can be amplified into the high optical purities observed in certain peptide‐forming systems. The authors construct a kinetic model based on reversible, stepwise, isodesmic polymerization in which L‑ and D‑amino‑acid monomers can add to or dissociate from both homochiral and heterochiral chain ends. The model is deliberately simple: it assumes a single forward rate constant (k) for monomer attachment, a single reverse constant (k′) for detachment, and a constant equilibrium constant K = k/k′ that is independent of chain length. The only structural parameters are the maximum chain length N and the initial concentrations of the two enantiomers. Mass conservation is enforced explicitly, reflecting the closed‑system nature of the experiments.
Mathematically, the system is described by a set of coupled ordinary differential equations for the concentrations of every possible polymer species up to length N. Each polymer is characterized by its composition (number of L and D residues) and by whether its terminal residue is L or D. Four elementary reactions are possible for each species: L‑addition to an L‑terminated chain, D‑addition to an L‑terminated chain, L‑addition to a D‑terminated chain, and D‑addition to a D‑terminated chain. Because the equilibrium constant is the same for all reactions, the model embodies the isodesmic hypothesis that the free‑energy change per monomer addition does not depend on the existing chain length or composition.
The authors fit the model to experimental data obtained by the Rehovot group, who studied chiral amplification of oligopeptides on well‑defined crystal lattices (e.g., Au(111), Cu(100)). In those experiments, a modest initial enantiomeric excess (often only a few percent) gave rise to a pronounced excess in specific oligomer lengths, a phenomenon the authors refer to as “lattice‑controlled chiral amplification.” Using a least‑squares optimization, the authors determined the best‑fit values of k and N for each chemical system. In all but one case, the model achieved correlation coefficients (R²) above 0.95, markedly superior to the random polymerization model (R²≈0.70). The outlier corresponded to a condition where temperature fluctuations or surface defects likely broke the assumption of uniform binding energy, highlighting the model’s sensitivity to the isodesmic premise.
A key insight emerging from the analysis is the identification of an autocatalytic‑like feedback embedded in the reversible kinetics. A tiny initial excess of one enantiomer biases the composition of the chain ends; because monomer addition is faster onto a chain end bearing the same chirality (due to the same equilibrium constant but a higher concentration of that monomer), subsequent steps preferentially extend homochiral sequences. This positive feedback becomes increasingly effective as the chain length grows, leading to a nonlinear, often exponential, increase in enantiomeric excess with N. The model therefore reproduces the experimentally observed steep rise in optical purity for longer oligomers without invoking external chiral catalysts or energy inputs.
The paper also discusses the thermodynamic consistency of the model. By enforcing detailed balance (k/k′ = K) and mass conservation, the authors ensure that the system evolves toward a well‑defined equilibrium distribution that depends only on K, N, and the initial total monomer concentrations. The fitted values of K are compatible with known peptide bond formation energies under the experimental conditions, lending further credibility to the approach.
In the discussion, the authors argue that the success of such a minimal model suggests that chiral amplification on surfaces can be driven primarily by kinetic selection rather than by specific catalytic asymmetry. They propose that the lattice geometry influences the effective K by modulating the binding orientation and steric constraints, thereby providing a physical basis for the observed “lattice control.” The single case where the model fails points to the need for extensions that incorporate non‑isodesmic effects (e.g., chain‑length‑dependent binding energies), multiple monomer types, or explicit solvent‑surface interactions.
Finally, the authors outline future directions: incorporating non‑isodesmic growth, exploring stochastic effects in very small systems, and applying the framework to prebiotic chemistry scenarios where surface‑mediated polymerization may have contributed to the emergence of biological homochirality. Overall, the study demonstrates that a reversible, isodesmic copolymerization model, despite its simplicity, captures the essential features of chiral amplification observed experimentally and provides a quantitative tool for predicting and designing enantioselective polymerization processes.
Comments & Academic Discussion
Loading comments...
Leave a Comment