MOLIERE: Automatic Biomedical Hypothesis Generation System
Hypothesis generation is becoming a crucial time-saving technique which allows biomedical researchers to quickly discover implicit connections between important concepts. Typically, these systems operate on domain-specific fractions of public medical…
Authors: Justin Sybr, t, Michael Shtutman
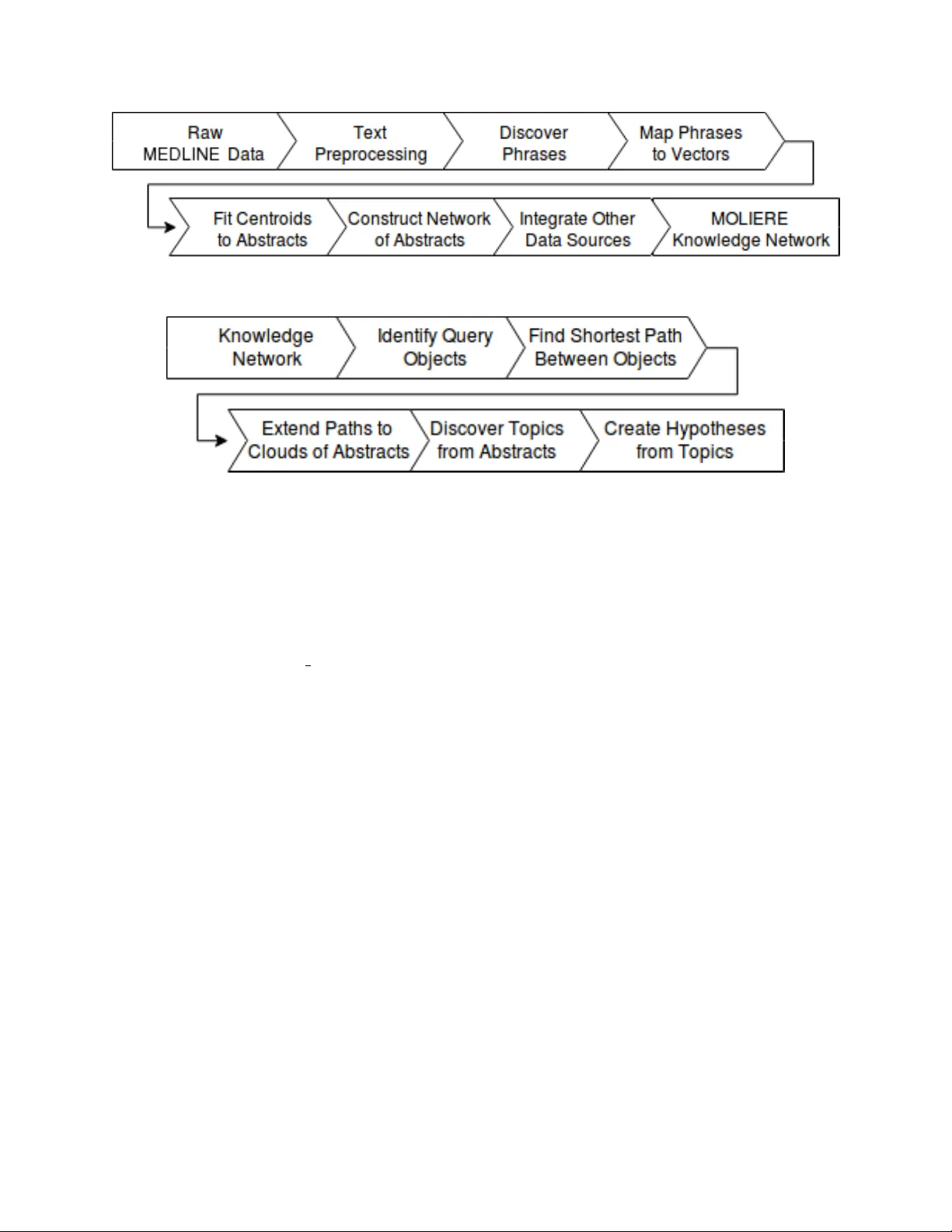
MOLIERE : Automatic Biomedical Hypothesis Generation System Justin Sybrandt 1 , Michael Shtutman 2 , Ilya Safro 1 1- Clemson Uni versity , School of Computing, Clemson SC, USA 2- Uni versity of South Carolina, Drug Disco very and Biomedical Sciences, Columbia SC, USA October 12, 2018 Abstract Hypothesis generation is becoming a crucial time-saving technique which allo ws biomedical re- searchers to quickly disco ver implicit connections between important concepts. T ypically , these systems operate on domain-specific fractions of public medical data. MOLIERE , in contrast, utilizes information from over 24.5 million documents. At the heart of our approach lies a multi-modal and multi-relational network of biomedical objects extracted from se v eral heterogeneous datasets from the National Center for Biotechnology Information (NCBI). These objects include but are not limited to scientific papers, keyw ords, genes, proteins, diseases, and diagnoses. W e model hypotheses using Latent Dirichlet Allo- cation applied on abstracts found near shortest paths discovered within this network, and demonstrate the effecti veness of MOLIERE by performing hypothesis generation on historical data. Our network, implementation, and resulting data are all publicly av ailable for the broad scientific community . 1 Intr oduction V ast amounts of biomedical information accumulate in modern databases such as MEDLINE [3], which currently contains the bibliographic data of ov er 24.5 million medical papers. These ever -gro wing datasets impose a great dif ficulty on researchers trying to survey and e valuate ne w information in the existing biomed- ical literature, e v en when advanced ranking methods are applied. On the one hand, the vast quantity and di versity of av ailable data has inspired many scientific breakthroughs. On the other hand, as the set of search- able information continues to grow , it becomes impossible for human researchers to query and understand all of the data rele vant to a domain of interest. In 1986 Swanson hypothesized that novel discov eries could be found by carefully studying the existing body of scientific research [45]. Since then, many groups hav e attempted to mine the wealth of public kno wledge. Ef forts such as Swanson’ s o wn Arrowsmith generate hypotheses by finding concepts which implicitly link two queried keyw ords. His method and others are discussed at length in Section 1.3. Ideally , an effecti v e hypothesis generation system greatly increases the producti vity of researchers. For example, imagine that a medical doctor belie ved that stem cells could be used to repair the damaged neural pathways of strok e victims (as some did in 2014 [22]). If no e xisting research directly linked stem cells to strok e victims, this doctor would typically ha ve no choice but to follow his/her intuition. Hypothesis generation allo ws this researcher to quickly learn the likelihood of such a connection by simply running a query . Our hypothetical doctor may query the topics stem cells and str oke for example. If the system returned topics such as paralysis then not only would the doctor’ s intuition be v alidated, b ut he/she would be more likely 1 to in vest in exploring such a connection. In this manner, an intelligent hypothesis generation system can increase the likelihood that a researcher’ s study yields usable new findings. 1.1 Our Contribution W e introduce a deployed system, MOLIERE [47], with the goal of generating more usable results than pre viously proposed hypothesis generation systems. W e de velop a novel method for constructing a large network of public knowledge and devise a query process which produces human readable text highlighting the relationships present between nodes. T o the best of our knowledge, MOLIERE is the first hypothesis generation system to utilize the entire MEDLINE data set. By using state-of-the-art tools, such as T oPMine [16] and F astT e xt [9], we are able to find novel hypotheses without restricting the domain of our knowledge network or the resulting vocab ulary when creating topics. As a result, MOLIERE is more generalized and yet still capable of identifying useful hypotheses. W e pro vide our network and findings online for others in the scientific community [47]. Additionally , to aid interested biomedical researchers, we supply an online service where users can request specific query results at http://jsybran.people.clemson.edu/mForm.php . Furthermore, MOLIERE is en- tirely open-source in order to facilitate similar projects. See https://github.com/JSybrandt/ MOLIERE for the code needed to generate and query the MOLIERE kno wledge network. In the follo wing paper we describe our process for creating and querying a large kno wledge network built from MEDLINE and other NCBI data sources. W e use natural language processing methods, such as Latent Dirichlet Allocation (LD A) [8] and topical phrase mining [16], along with other data mining techniques to conceptually link together abstracts and biomedical objects (such as biomedical ke ywords and n -grams) in order to form our network. Using this netw ork we can run shortest path queries to discover a pathway between two concepts which are non-trivially connected. W e then find clouds of documents around these pathways which contain knowledge representative of the path as a whole. PLDA+, a scalable implementation of LD A [28], allo ws us to quickly find topic models in these clouds. Unlike similar systems, we do not restrict PLD A+ to any set vocabulary . Instead, by using topical phrase mining, we identify meaningful n-grams in order to improve the performance, flexibility , and understandability of our LD A models. These models result in both quantitativ e and qualitativ e connections which human researchers can use to inform their decision making. W e e valuate our system by running queries on historical data in order to disco ver landmark findings. For e xample, using data published on or before 2009, we find strong e vidence that the protein Dead Box RN A Helicase 3 (DDX3) can be applied to treat cancer . W e also verify the ability of MOLIERE to make predictions similar to pre vious systems with restricted LD A [49]. 1.2 Our Method in Summary W e focus on the domain of medicine because of the large wealth of public information provided by the National Library of Medicine (NLM). MEDLINE is a database containing over 24.5 million references to medical publications dating all the way back to the late 1800s [3]. Over 23 million of these references include the paper’ s title and abstract text. In addition to MEDLINE, the NLM also maintains the Unified Medical Language System (UMLS) which is comprised of three main resources: the metathesaurus, the semantic network, and the SPECIALIST natural language processing (NLP) tools. These resources, along with the rest of our data, are described in section 2.1. 2 Our kno wledge base starts as XML files provided by MEDLINE, from which we extract each publica- tion’ s title, document ID, and abstract text. W e first process these results with the SPECIALIST NLP toolset. The result is a corpus of text which has standardized spellings (for example “colour” becomes “color”), no stop words (including medical specific stop words such as Not Otherwise Specified (NOS) ), and other char- acteristics which improv e later algorithms on this corpus. Then we use T oPMine to identify multi-word phrases from that corpus such as “asthma attack, ” allowing us to treat phrases as single tokens [16]. Ne xt, we send the corpus through F astT e xt , the most recent word2vec implementation, which maps each unique token in the corpus to a vector [30]. W e can then fit a centroid to each publication and use the Fast Library for Approximate Nearest Neighbors (FLANN) to generate a nearest neighbors graph [32]. The result is a network of MEDLINE papers, each of which are connected to other papers sharing a similar topic. This network, combined with the UMLS metathesaurus and semantic network, constitutes our full kno wledge base. The network construction process is described in greater detail in Section 2. W ith our netw ork, a researcher can query for the connections between two keyw ords. W e find the short- est path between the two keywords in the knowledge network, and extend this path to identify a significant set of related abstracts. This subset contains many documents which, due to our network construction pro- cess, all share common topics. W e perform topic modeling on these documents using PLD A+ [28]. The result is a set of plain te xt topics which represent dif ferent concepts which likely connect the two queried ke ywords. More information about the query process is detailed in Section 3. W e use landmark historical findings in order to validate our methods. For example, we show the implicit link between V enlafaxine and HTR1A, and the inv olvement of DDX3 on Wnt signaling. These queries and results are detailed in Section 4. In Sections 5 and 6 we discuss challenges and open research questions we hav e uncov ered during our work. 1.3 Related W ork The study and exploration of undiscov ered public kno wledge be gan in 1986 with Swanson’ s landmark paper [45]. Swanson hypothesized that fragments of information from the set of public knowledge could be connected in such a w ay as to shed light on ne w discov eries. W ith this idea, Swanson continued his research to dev elop Arrowsmith , a text-based search application meant to help doctors make connections from within the MEDLINE data set [38, 44, 46]. T o use Arro wsmith , researchers supply two UMLS keyw ords which are used to find tw o sets of abstracts, A and C . The system then attempts to find a set B ≈ A ∩ C . Assuming sets A and C do not o verlap initially , implicit textual links are used to expand both sets until some sizable set B is discov ered. The experimental process was computationally expensiv e, and queries were typically run on a subset of the MEDLINE data set (according to [46] around 1,000 documents). Spangler has also been a driving force in the field of hypothesis generation and mining undisco vered public kno wledge. His te xtbook [41] details many text mining techniques as well as an e xample application related to hypothesis generation in the MEDLINE data set. His research in this field has focused on p53 kinases and how these undisco vered interactions might aid drug designers [42, 41]. His method le verages unstructured text mining techniques to identify a network entities and relationships from medical text. Our work dif fers from this paradigm by utilizing the structured UMLS keyw ords, their known connections, and mined phrases. W e do, howe ver , rely on similar unstructured text mining techniques, such as F astT e xt and FLANN, to make implicit connections between the abstracts. Rzhetsky and Evans notice that current information gathering methods struggle to keep up with the gro wing wealth of forgotten and hard to find information [17]. Their work in the field of hypothesis gener- ation has included a study on the assumptions made when constructing biomedical models [15] and digital representations of hypothesis [40]. 3 Di voli et al. analyze the assumptions made in medical research [15]. The y note that scientists often reach contradictory conclusions due to differences in each person’ s underly assumptions. The study in [15] highlights the variance of these preconceptions by surveying medical researchers on the topic of cancer metastasis. Surprisingly , 27 of the 28 researchers surv eyed disagree with the textbook process of cancer metastasis. When asked to provide the “correct” metastasis scenario, none of the surve yed scientists agree. Di voli’ s study highlights a major problem for hypothesis generation. Scientists often disagree, ev en in published literature. Therefore, a hypothesis generation system must be able to produce reliable results from a set of contradicting information. In [40], Soldatova and Rzhetsky describe a standardized way to represent scientific hypotheses. By creating a formal and machine readable standard, they en vision a collection of h ypotheses which clearly describes the full spectrum of existing theories on a giv en topic. Soldato v a and Rzhetsky extend e xisting approaches by representing hypotheses as logical statements which can be interpreted by Adam , a robot scientist capable of starting one thousand experiments a day . Adam is successful, in part, because they model hypotheses as an ontology which allo ws for Bayesian inference to gov ern the likelihood of a specific hypothesis being correct. DiseaseConnect, an online system that allows researchers to query for concepts intersecting two key- words, is a notable contrib ution to hypothesis generation [27]. This system, proposed by Liu et al., is similar to both our system and Arrowsmith [39] in its focus on UMLS keyw ords and MEDLINE literature min- ing. Unlike our system, Liu et al. restrict DiseaseConnect to simply 3 of the 130 semantic types. They supplement this subset with concepts from the OMIM [19] and GW AS [6] databases, tw o genome specific data sets. Still, their network size is approximately 10% of the size of MOLIERE . DiseaseConnect uses its network to identify diseases which can be grouped by their molecular mechanisms rather than symptoms. The process of finding these clusters depends on the relationships between dif ferent types of entities present in the DiseaseConnect network. Users can vie w sub-networks relev ant to their query online and related entities are displayed alongside the network visualization. Barab ´ asi et al. improve upon the network analytic approach to understand biomedical data in both their work on the disease network [19] as well as their more generalized symptoms-disease network [51]. In the former [19], the authors construct a bipartite network of disease phonemes and genomes to which they refer to as the Diseasome . Their inspiration is an observ ation that genes which are related to similar disorders are likely to be related themselv es. They use the Diseasome to create two projected networks, the human disease network (HDN), and the Disease Gene Network (DGN). In the latter [51], the y construct a more generalized human symptoms disease network (HSDN) by using both UMLS keyw ords and bibliographic data. HSDN consists of data collected from a subset of MEDLINE consisting of only abstracts which contained at least one disease as well as one symptom, a subset consisting of approximately 850,000 records. From this set, Goh et al. calculated keyw ord co-occurrence statistics in order to build their network. They v alidate their approach using 1,000 randomly selected MEDLINE documents and, with the help of medical experts, manually confirm that the relationship described in a document is reflected meaningfully in HSDN. Ultimately , Goh et al. find strong correlations between the sympt oms and genes shared by common diseases. Bio-LD A is a modification of LD A which limits the set of ke ywords to the set present in UMLS [49]. This reduction improv es the meaning and readability of topics generated by LDA. W ang et al. also show in this work that their method can imply connections between keywords which do not show up in the same document. For example, they note that V enlafaxine and HTR1A both appear in the same topic ev en though both do not appear in the same abstract. W e explore and repeat these findings in Section 4.2. 4 1.4 Related and Incorporated T echnologies FastT ext is the most recent implementation of word2vec from Milkolov et al. [30, 31, 23, 9]. W ord2vec is a method which utilizes the skip-gram model to identify the relationships between words by analyzing word usage patterns. This process maps plain text words into a high dimensional vector space for use in data mining applications. Similar w ords are often grouped together , and the distances between words can rev eal relationships. F or example, the distance between the words “Man” and “W oman” is approximately the same as the distance between “King” and “Queen”. F astT e xt improv es upon this idea by le veraging sub-strings in long rarely occurring words. T oPMine , a project from El-Kishky et al., is focused on discov ering multi-word phrases from a lar ge corpus of text [20]. This project intelligently groups unigrams together to create n-gram phrases for later use in text mining algorithms. By using a bag-of-words topic model, T oPMine groups unigrams based on their co-occurrence rate as well as their topical similarity using a process they call Phrase LD A. Latent Dirichlet Allocation [8] is the most common topic modeling process and PLDA+ is a scalable implementation of this algorithm [20, 28]. Dev eloped by Zhiyuan Liu et al., PLD A+ quickly identifies groups of words and phrases which all relate to a similar concept. Although it is an open research question as to how best to interpret these results, simple qualitativ e analysis allows for “ballpark” estimations. For instance, it may take a medical researcher to wholly understand the topics generated from abstracts related to two keywords, b ut an yone can identify that all words related to a concept of interest occur in the same topic. Results like this, sho w that LDA has distinguished the presence of a concept in a body of te xt. 2 Knowledge Netw ork Construction In order to discover hypotheses we construct a large weighted multi-layered network of biomedical objects extracted from NLM data sets. Using this network, we run shortest-centroid-path queries (see Section 3) whose results serve as an input for h ypothesis mining. The wall clock time needed to complete this network construction pipeline is depicted in Figure 1 (see details in Section 4.4 ). Omitted from this figure is the time spent preprocessing the initial abstract text due to its embarrassingly parallel nature. 2.1 Data Sources The NLM maintains multiple databases of medical information which are the main source of our data. This includes MEDLINE [3], a source containing the metadata of approximately 24.5 million medical publica- tions since the late 1800’ s. Most of these MEDLINE re cords include a paper’ s title, authors, publication date, and abstract text. In addition to MEDLINE, the NLM maintains UMLS [2], which in turn provides the metathesaurus as well as a semantic network. The metathesaurus contains two million k eywords along with all kno wn syn- onyms (referred to as “atoms”) used in medical text. For example, the keyword “RN A ” has many different synonyms such as “Ribonucleinicum acidum”, “Ribonucleic Acid”, and “Gene Products, RN A ” to name a fe w . These metathesaurus ke ywords form a network comprised of multi-typed edges. F or example, an edge may represent a par ent - child or a boarder concept - narr ower concept relationship. RN A has connections to terms such as “Nucleic Acids” and “DN A Synthesizers”. Lastly , each keyword holds a reference to an ob- ject in the semantic network. RNA is an instance of the “Nucleic Acid, Nucleoside, or Nucleotide” semantic type. The UMLS semantic network is comprised of approximately 130 semantic types and is connected in a similar manner as the metathesaurus. For example, the semantic type “Drug Deli very De vice” has an “is 5 Figure 1: Running times of each network construction phase. All phases run on a single node described in section 4.4. Not sho wn: Initial text processing which w as handled by a large array of small nodes. a” relationship with the “Medical Device” type, and has a “contains” relationship with the “Clinical Drug” type. MEDLINE, the metathesaurus, and the semantic network are represented in our network as dif ferent layers. Articles which contain full text abstracts are represented as the abstract layer nodes A , k eyw ords from the metathesaurus are represented as nodes in the keyword layer K , and items from the semantic network are represented as nodes in the semantic layer S . 2.2 Network T opology W e define a weighted undirected graph underlying our network N as G = ( V , E ) , where V = A ∪ K ∪ S . The construction of G was gov erned by two major goals. Firstly , the shortest path between tw o indirectly related k eyw ords should likely contain a significant number of nodes in A . If instead, this shortest path contained only K − K edges, we would limit ourselves to kno wn information contained within the UMLS metathesaurus. Secondly , conceptual distance between topics should be represented as the distance between two nodes in N . This implies that we can determine the similarity between i, j ∈ V by the weight of their shortest path. If ij ∈ E , this w ould imply that exists a pre viously known relationship between i and j . W e are instead interested in connections between distant nodes, as these potentially represent unknown information. Belo w we describe the construction of each layer in N . 2.3 Abstract Layer A When connecting abstracts ( A − A edges), we want to ensure that two nodes i, j ∈ A with similar content are likely neighbors in the A layer . In order to do this, we turned to the UMLS SPECIALIST NLP toolset [1] as well as T oPMine [16] and F astT e xt [9, 23]. Our process for constructing A is summarized in Figure 2. 6 Figure 2: MOLIERE network construction pipeline. Figure 3: MOLIERE query pipeline. First, we extract all titles, abstracts, and associated document ID (referred to as PMID within MEDLINE) from the raw MEDLINE files. W e then process these combined titles and abstracts with the SPECIALIST NLP toolset to standardize spelling, strip stop words, con vert to ASCII, and perform a number of other data cleaning processes. W e then use T oPMine to generate meaningful n -grams and further clean the text. This process finds tokens that appear frequently together , such as newborn and infants and combines them into a single token newborn infants . Cleaning and combining tokens in this manner greatly increases the performance of F astT e xt , the next tool in our pipeline. When running T oPMine , we keep the minimum phrase frequency and the maximum number of words per phrase set to their def ault values. W e also keep the topic modeling component disabled. On our a v ailable hardware, the MEDLINE data set can be processed in approximately thirteen hours without topic modeling, but does not finish within three days if topic modeling is enabled. Because the resulting phrases are of high quality e ven without the topic modeling component, we accept this quality vs. time trade off. It is also important to note that we modify the version of T oPMine distributed by El-Kishky in [16] to allow phrases containing numbers, such as gene names like p53. Next, F astT e xt maps each token in our corpus to a v ector v ∈ R d , allowing us to fit a centroid per abstract i ∈ A . Using a sufficiently high-dimensional space ensures a good separation between vectors. In other w ords, each abstract i ∈ A is represented in R d as c i = 1 /k · P k j =1 x j , where x j are F astT ext vectors of k keyw ords in i . W e choose to use the skipgram model to train F astT e xt and reduce the minimum word count to zero. Because our data preprocessing and T oPMine hav e already stripped low support words, we accept that any n-gram seen by F astT e xt is important. Follo wing examples presented in [30, 31, 23] and others, we set the dimensionality of our vector space d to 500. This is consistent with published examples of similar size, for example the Google news corpus processed in [30]. Lastly , we increase the word neighborhood and number of possible sub-words from fi ve to eight in order to increase data quality . Finally , we used FLANN [32] to create nearest neighbors graph from all i ∈ A in order to establish 7 A − A edges in E . This requires that we presuppose a number of expected nearest neighbors per abstract k . W e set this tunable parameter to ten initially and noticed that this v alue seemed appropriate. By studying the distances between connected abstracts, we observed that most abstracts had a range of very close and relati vely far “nearest neighbors”. For our purposes in these initial e xperiments, we kept k = 10 and saw promising results. Due to time and resource limitations, we were unable to explore higher values of k in this study , but we are currently planning experiments where k = 100 and k = 1000 . It is important to note that the resulting network will hav e ≈ k (2 . 3 × 10 7 ) edges, so there is a considerable trade-off between quality vs. space and time complexity . After experimenting with both L 2 and normalized cosine distances, we observed that L 2 distance met- ric performs significantly better for establishing connections between centroids. Unfortunately , we cannot utilize the k-tree optimization in FLANN along with non-normalized cosine distance, making it computa- tionally infeasible a dataset of our size. This is because the k-tree optimization requires an agglomerati ve distance metric. Lastly , we scale edges to the [0 , 1] interv al in order to relate them to other edges within the network. 2.4 K eyword Layer K The K layer is imported from the UMLS metathesaurus. Each keyword is referenced by a CUI number of UMLS. This layer links keyw ords which share already kno wn connections. These kno wn connections are K − K edges. The metathesaurus connections link related words; for example, the keyword “Protine p53” C0080055 is related to “T umor Suppressor Proteins” C0597611 and “Li-Fraumeni Syndrome” C0085390 among others. There exist 14 different types of connections between ke ywords representing relationships such as par ent - child or br oader concept - narr ower concept . W e assign each a weight in the [0 , 1] interval corresponding to its relev ance, and then scale all weights by a constant factor σ so the average A − A edge are is stronger than the av erage K − K edge. The result is that a path between two indirectly related concepts will more likely include a number of abstracts. W e selected σ = 2 , b ut more study is needed to determine the appropriate edge weights within the ke yword layer . 2.5 A − K Connections In order to create edges between A and K , we used a simple metric of term frequency-in verse document frequency (tf-idf). UMLS provides not only a list of ke ywords, but all kno wn synonyms for each key- word. For example, the ke yword Color C0009393 has the American spelling, the British spelling, and the pluralization of both defined as synonyms. Therefore we used the raw text abstracts and titles (before run- ning the SPECIALIST NLP tools) to calculate tf-idf. In order to quickly count all occurrences of UMLS ke ywords across all synon yms, we implemented a simple parser . This was especially important because many keywords in UMLS are actually multi-word phrases such as “Clustered Regularly Interspaced Short Palindromic Repeats” (a.k.a. CRISPR) C3658200 . In order to count these keyw ords, we construct a parse tree from the set of synonyms. Each node in the tree contains a word, a set of CUIs, and a set of children nodes, with the e xception of the root which contains the null string. W e b uild this tree by parsing each synonym word by word. For each word, we either create a new node in the tree, or traverse to an already existing child node. W e store each synonym’ s CUI in the last node in its parse path. Then, to parse a document, we simply trav erse the parse tree. This can be done in parallel ov er the set of abstracts. For each word in an abstract, we move from the current tree node to a child representing the same word. If none exists, we return to the root node. At each step of this traversal, we record the CUIs present at each visited node. In this manner , we get a count of each CUI 8 present in each abstract. Our ne xt pass aggregates these counts to disco ver the total number of usages per ke yword across all abstracts. W e calculate tf-idf per ke yword per abstract. Because our network’ s weights represent distance, we take the in verse of tf-idf to find the weight for an A − K edge. This is done simply by di viding a CUI’ s count across all abstracts by its count in a particular abstract. By calculating weights this way , abstracts which use a keyw ord more often will have a lower weight, and therefore, a shorter distance. W e scale the edge weights to the [0 , σ ] interval so that these edges are comparable to those within the A and K layers. 2.6 Semantic Layer S The UMLS supplies a companion network referred as the semantic network. This network consists of semantic types, which are overarching concepts. These “types” are similar to the function of a “type” in a programming language. In other words, it is a conceptual entity embodied by instantiations of that type. In the UMLS network, elements of K are analogous to the instantiations of semantic types. While there are ov er tw o million elements of K , there are approximately 130 elements in S . For e xample, the semantic type Disease or Syndrome T047 is defined as “ A condition which alters or interferes with a normal process, state, or acti vity of an or ganism” [2]. There are thousands of keyw ords, such as “influenza” C0021400 that are instances of this type. The S − S edges are connected similarly to K − K edges. The overall structure is hierarchical with “Event” T051 and “Entity” T071 being the most generalized semantic types. Cross cutting connections are also present and can tak e on approximately fifty dif ferent forms. These cross cutting relations also form a hierarchy of relationship types. For example, “produces” T144 is a more specific relation than its parent “brings about” T187 . W e initially included S in our network by linking each keyword to its corresponding semantic type. Unfortunately , in our early results we found that man y shortest paths tra versed through S rather than through A . For e xample, if we were interested in two diseases, it was possible for the shortest path w ould simply trav el to the “Disease or Syndrome” T047 type. This ultimately degraded the performance of our hypothesis generation system. As a result we remov ed this layer , but that further study may find that careful choice of S − S and K − S connection weights may mak e S more useful. This is further discussed in Section 5. 3 Query Pr ocess The process of running a query within MOLIERE is summarized in Figure 3. Running a query starts with the user selecting two nodes i, j ∈ V (typically , but not necessarily , i, j ∈ K ). F or example, a query searching for the relationship between “stem cells” and “strokes” would be input as ke yword identifiers C0038250 and C1263853 , respecti vely . This process simplifies our query process, but determining a lar ger set of ke ywords and abstracts which best represents a user’ s search query is a future work direction. After recei ving two query nodes i and j , we find a shortest path between them, ( ij ) s , using Dijkstra’ s algorithm. These paths typically are between three and fi ve nodes long and contain up to three abstracts (unless the nodes are truly unrelated, see Section 4.1). W e observed that when ( ij ) s contains only two or three nodes in K , that the ij relationship is clearly well studied because it w as solely supplied by the UMLS layer K . W e are more interested in paths containing abstracts because these represent keyw ord pairs whose relationships are less well-defined. Still, the abstracts we find along these shortest paths alone are not likely to be suf ficient to generate a hypothesis. 9 Figure 4: Process of extending a path to a cloud of abstracts. 3.1 Hypothesis Modeling Broadening ( ij ) s consists of tw o main phases, the results of which are depicted in Figure 4. First, we select all nodes S = ( ij ) s ∩ A . These abstracts along the path ( ij ) s represent papers which hold key informa- tion relating tw o unconnected ke ywords. W e find a neighborhood around S using a weighted breadth-first trav ersal, selecting the closest 1,000 abstracts to S . W e will call this set N . Because A w as constructed as a nearest neighbors graph, it is lik ely that the concepts contained in N will be similar to the concepts contained in S , which increases the likelihood that important concepts will be detected by PLDA+ later in the pipeline. Next, we identify abstracts with contain information pertaining to the K − K connections present in ( ij ) s . W e do so in order to identify abstracts which likely contain concepts which a human reader could use to understand the kno wn relationship between two connected k eyw ords. W e start by trav ersing ( ij ) s to find α, β ∈ K such that α and β are adjacent in ( ij ) s . From there, we find a set of abstracts C = { c : cα ∈ E ∧ cβ ∈ E } . That is, C is a subset of abstracts containing both k eywords α and β . Because ( ij ) s can ha ve many edges between keywords, and because thousands of abstracts can contain the same two keywords, it is important to limit the size of C . This process creates a set of around 1,300 A -nodes. This set will typically contain around 15,000-20,000 words and is large enough for PLD A+ to find topics. W e run PLD A+ and request 20 topics. W e find this provides a suf ficient spread in our resulting data sets. The trained model generated by PLD A+ is what is e ventually returned by our query process. For our experiments, we often must process tens of thousands of results and thus must train topic models quickly . This is most apparent when running a one-to-many query such as the drug repurposing example in 4.3. Additionally , the training corpus returned from a MOLIERE query is often only a couple thousand documents lar ge. As a result, we set the number of topics and the number of iterations to relati vely small v alues, 20 and 100 respectively . Because we store intermediary results, it is trivial to retrain a topic model if the preliminary result seems promising. The process of analyzing a topic model and uncovering a human interpretable sentence to describe a hypothesis is still a pressing open problem. The process as stated here does have some strong benefits which are apparent in Section 4. These include the ability to find correlations between medical objects, such as between a drug and multiple genes. In Section 6 we explain our initial plans to improv e the quality of results which can be deduced from these topic models. 10 4 Experiments W e conduct tw o major validation ef forts to demonstrate our system’ s potential for hypothesis generation. For each of these e xperiments we use the same set of parameters for our trained model and netw ork weights. Our initial findings sho w our choices, detailed in Section 2, to be robust. W e plan to refine these choices with methods described in Section 6. W e repeat an e xperiment done by W ang et al. in [49] wherein we disco ver the implicit connections between the drug V enlafaxine and the genes HTR1A and HTR2A. W e also perform a lar ge scale study of Dead Box RNA Helicase 3 (DDX3) and its connection to cancer adhesion and metastasis. Each of these experiments is described in greater detail in the following sections. In this paper , we deliberately do not evaluate our e xperiments with extr emely popular objects such as p53. These objects ar e so highly connected within K that hypothesis generation in volving these ke ywor ds is easy for many differ ent methods. 4.1 Network Pr ofile W e conduct our e xperiments on a v ery large knowledge graph which has been constructed according to Sec- tion 2. W e initially created a network N containing information dating up to and including 2016. This net- work consists of 24,556,689 nodes and 989,169,295 edges. The network overall consists of lar gest strongly connected component containing 99.8% of our network. The average degree of a node in N is 79.65, and we observe a high clustering coef ficient of 0.283. These metrics cause us to e xpect that the shortest path between tw o nodes will be v ery short. Our experiments agree, showing that most shortest paths are between three and six nodes long. 4.2 V enlafaxine to HTR1A W ang et al. in [49] use a similar topic modeling approach, and find during one of their e xperiments that V en- lafaxine C0078569 appears in the same topic as the HTR1A and HTR2A genes ( C1415803 and C1825553 respecti vely). When looking into these results, they find a stronger association between V enlafaxine and HTR1A. This finding is important because V enlafaxine is used to treat depressiv e disorder and anxiety , which HTR1A and HTR2A have been thought to af fect, but as of 2009 no abstract contains this link. As a result, this implicit connection is dif ficult to detect with many e xisting methods. Results: As a result of running tw o queries, V enlafaxine to HTR1A, and V enlafaxine to HTR2A, we can corroborate the findings of W ang et al. in [49]. W e find that neither pair of keyw ords is directly con- nected or connected through a single abstract. Nev ertheless, phrases such as “long term antidepressant treatment, ” “action antidepressants, ” and “antidepressant drugs” are all prominent ke ywords in the HTR1A query . Meanwhile, the string “depress” only occurs four times in unrelated phrases with the HTR2A results. The distribution of depression related k eyw ords from both queries can be see in figure 5. Similarly , our results for HTR1A contain a single topic holding the phrases “anxiogenic, ” “anxiety disorders, ” “depression anxiety disorders, ” and “anxiolytic response. ” In contrast, our HTR2A results do not contain any phrases related to anxiety . The distribution of anxiety related keyw ords from both queries can be see in figure 6. Our findings agree with those of W ang et al. which were that a small association score of 0.34 between V enlafaxine and HTR1A indicates a connection which is likely related to depressi ve disorder and anxiety . The association score between V enlafaxine and HTR2A, in contrast, is a much higher 4.0. This indicates that the connection between these two ke ywords is much weak er . 11 Figure 5: Distrib ution of n-grams having to do with depression from V enlaf axine queries. Figure 6: Distrib ution of n-grams having to do with anxiety from V enlaf axine queries. 12 4.3 Drug Repurposing and DDX3’ s Anti-T umor Applications Many genes are active in multiple cellular processes and in man y cases they are found to be active outside of the original area in which the gene was initially discovered. The prediction of ne w processes is especially important for repurposing existing drugs (or drug target genes) to a new application [5, 33, 4]. As an example, the drugs developed for the treatment of infectious diseases were recently repurposed for cancer treatment. Extending applications of e xisting drugs provides a tremendous opportunity for the de velopment of cost-ef fectiv e treatments for cancers and other life-threatening diseases. T o estimate the predictiv e v alue of our system for the discovery of new applications of small molecules we select Dead Box RN A Helicase 3 (DDX3) C2604356 . DDX3 is the member of Dead-box RN A helicase and was initially discov ered to be a re gulator of transcription and propagation of Human Immunodeficienc y V irus (HIV) as well as ribosomal biogenesis. Initially , DDX3 was a target for the development of anti-viral therapy for the AIDS treatment [25, 29]. More recently , DDX3 acti vity w as found to be in volv ed cancer de velopment and progression mainly through regulation of the Wnt signaling pathway [13, 50] and associated regulation of Cell-cell and Cell- matrix adhesion, tumor cells in vasion, and metastasis [12, 43, 48, 24]. Currently , DDX3 is an established target for anti-tumor drug de velopment [10, 37, 11] and represents a case for repurposing target anti-viral drugs into the application area of anti-tumor therapy . T o test this hypothesis, we analyze the data a v ailable on and before 12/31/2009, when no published indication of links in between DDX3 and the Wnt signaling were av ailable. W e compare DDX3 to all UMLS ke ywords containing the te xt “signal transduction”, “transcription”, “adhesion”, “cancer”, “dev elopment”, “translation”, or “RN A” in their synonym list. This search results in 9,905 keywords over which we query for relationships to DDX3. From this large set of results we personally analyze a subset of important pairs. Results: In our generated dataset, we found follo wing text grouping within topics: “substrate adhesion, ” “RGD cell adhesion domain, ” “cell adhesion factor , ” “focal adhesion kinase” which are indicative for the cell-matrix adhesion. The topics “cell-cell adhesion, ” “regulation of cell-cell adhesion, ” “cell-adhesion molecules” indicate the in volvement of DDX3 into cell-cell adhesion regulation. The inv olvement of adhe- sion is associated with topics related to tumor dissemination: “ Collaborati ve staging metastasis ev aluation Cancer , ” “metastasis adhesion protein, human, ” “metastasis associated in colon cancer 1” (selected in be- tween others similar topics). The results abov e suggested that through analysis of the ≤ 2009 dataset we can predict the inv olvement of DDX3 in tumor cell dissemination through the ef fects of Cell-cell and cell-matrix adhesion. Next, we analyzed, whether it will be possible to made inside of the mechanisms of DDX3-dependent regulation of Wnt signaling. As sho wn recently , DDX3 in v olvement on Wnt signaling is based on the re gulated Casein kinase epsilon, to affect phosphorylation of the dishe veled protein. Although we cannot predict the exact mechanism of DDX3 based on ≤ 2009 dataset, the existence of multiple topics of signal-transduction asso- ciated kinases, like “CELL ADHESION KINASE”, “activ ation by or ganism of defense-related host MAP kinase-mediated signal transduction pathway”, “modulation of defense-related symbiont mitogen-activ ated protein kinase-mediated signal transduction pathway by organism”, suggested the ability of DDX3 to regu- late kinases acti vities and kinase-regulated pathways. 4.4 Experimental Setup W e performed all experiments on a single node within Clemson’ s Palmetto supercomputing cluster . T o perform our experiments and construct our network, we use an HP DL580 containing four Intel Xeon x7542 chips. This 24 core node has 500 GB of memory and access to a large ZFS-based file system where we 13 stored experimental data. For the DDX3 queries, we initially searched for all ( ij ) s where i = DDX3 and j ∈ K . This resulted in 1,350,484 shortest paths with corresponding abstract clouds. W e used PLD A+ to construct models for all of these paths. Discovering all ( ij ) s completed in almost 10 hours of CPU time, and training the respectiv e models completed in slightly ov er 68 hours of CPU time. W e ran PLD A+ in parallel, resulting in a wall time of only 12 hours. As mentioned pre viously , this large dataset w as filtered to the 9,905 paths we are interested in. W e generate the results for the V enlafaxine e xperiments in one hour of CPU time, which is mostly spent loading our very large network and then running Dijkstra’ s algorithm. After this, the two resulting PLD A+ models were trained in parallel within a minute. 5 Deployment Challenges In the follo wing section we detail the challenges which we ha ve faced and are e xpecting to encounter while creating our system and deploying it to the research community . Dynamic Information Updates The process of creating our network is computationally expensiv e and for the purposes of validation we must create multiple instances of our network representing different points in time. Initially we would hav e liked to create these multiple instances from scratch, starting from the MED- LINE archiv al distribution and rebuilding the network from there. Unfortunately , this prov ed infeasible because creating a single network is a time consuming process. Instead, we filter our network by removing abstracts and ke ywords which were published after our select date. Additionally , the act of adding informa- tion to our network, such as extending the 2016 network to create a 2017 network, is not straightforward. Ideally , adding a small number abstracts or k eywords should be a fast and dynamic process which only af fects localized regions of the network. If this were so, our deployed system could take advantage of new ideas and connections as soon as they are published. A deployed system could support dynamic updates with an amortized approach. Using previously cre- ated F astT e xt and T oPMine models, ne w documents could be fitted into an existing network with suitably high performance. Of course, if a new document introduced a new ke yword or phrase, we would be unable to detect it initially . After some threshold of new documents had been added to the network, we could then rerun the entire network construction process to ensure that new k eyw ords, phrases, and concepts would be properly placed in the network. Query Platform and Perf ormance: Initially , we expected to use a graph database to make the query process easier . W e surve yed a selection of graph databases and found that Neo4j [14] provides a po werful query language as well as a platform capable of holding our billion-edge network. Unfortunately , Neo4j does not easily support weighted shortest path queries. Although some user suggestions did hint that it may be possible, the process requires le veraging edge labels and custom jav a procedures in a w ay that did not seem scalable. In place of Neo4j, we implemented Dijkstra’ s shortest path algorithm in C++ using ske w heaps as the internal priority queue. This implementation was chosen to minimize memory usage while maximizing speed and readability . Because we implemented Dijkstra’ s algorithm ourselves, we can also combine the process of finding a shortest path and finding all neighboring abstracts for all keywords from a specific source. W ith only these high lev el optimizations, we were able to generate over 1,350,000 shortest paths and abstract neighborhoods in under ten hours, b ut generating a single result takes slightly o ver one hour . 14 6 Lessons Learned and Open Pr oblems Specialized LD A: During last two decades there has been a number of significant attempts to design au- tomatic hypothesis generation systems [41, 44, 49]. Howe ver , most of these improve their performance by restricting either their information space or the size of their dictionary . For e xample, specialized v ersions of LD A such as Bio-LDA [49] uncover latent topics using a dictionary that gives a priority to special terms. W e find that such approaches are helpful when general language may significantly ov er weigh a specialized lan- guage. Howe ver , phrase mining approaches that recov er n -grams, such as [16], produce accurate methods without limiting the dictionary . Hypothesis V iability and Novelty Assessment: Intuitiv ely , a strong connection between two concepts in N means that there exist a significant amount of research that covers a path between them. Similar observ ations are v alid for LDA, i.e., latent topics are likely to describe well known facts. As a result, the most meaningful connections and interpretable topical inference are disco vered with latent keywords that are among the most well known concepts. Howe ver , real hypotheses are not necessarily described using the most latent keywords in such topic models. In many cases, the keywords required for a successful and interpretable hypothesis start to appear among 20-30 most latent topical keywords. Thus, a major open problem related is the process to which one should select a combination of k eyw ords and topics in order to represent a viable hypothesis. This problem is also linked to the problem of assessing the viability of a generated hypothesis. These problems, as well as the problem of hypothesis nov elty assessment, can be partially addressed by using the Dynamic T opic Modeling (DTM) [7]. Our preliminary experiments with scalable time-dependent clustered LD A [21] that significantly accelerates DTM demonstrate a potential to discover dynamic topics in MEDLINE. The dynamic topics are typically more realistic than those that can be disco vered in the static network. This significantly simplifies the assessment of viability and topic noise elimination. Incorporating the Semantic Layer S : In section 2.6 we describe the process in which we ev aluated the UMLS semantic network and found that it worsened our resulting shortest path queries. Further work could improve the contribution that S has on our overall network, possibly allowing S to define the ov erall structure of our knowledge graph. In order to do this, one would likely need to take into account the hierarchy of relationship types present in this network, as well as the relativ e relationship each element in K has with its connection in S . Ultimately , these different relationships would need to inform a weighting scheme that balances the over generalizations that S introduces. For e xample, it may be useful to understand that two ke ywords are both diseases, b ut it is much less useful to understand that two ke ywords are “entities”. Learning the Models of Hypothesis Generation: There is surprisingly little research focused on address- ing the process of biomedical research and how that process e v olves ov er time. W e w ould like to model the process of discovery formation, taking into account the information context surrounding and preceding a disco very . W e belie ve we could do so by re verse engineering existing discoveries in order to discover factors which altered the steps in a scientist’ s research pipeline. Se veral promising observ ations in this di- rection hav e been done by Foster et al. [18] who e xamined this through Bourdieu’ s field theory of science by de veloping a typology of research strategies on networks extracted from MEDLINE. Howe v er , instead of re verse engineering their models, they separate innov ation steps from those that are more traditional in the research pipeline. Dynamic Keyword Discovery: One of the limitations we found when performing our historical queries is the delay between the first major uses of a keyword and its appearance in the UMLS metathesaurus. Initially , we planned to study the relationship between “CRISPR” C3658200 and “genome editing” C4279981 . T o our surprise, many k eyw ords related to this query did not exist in our historical networks between 2009 15 and 2012, despite their frequent usage in cutting-edge research during that time. T o further confuse the issue, although the keyword “CRISPR” did not appear in the UMLS releases on or before 2012, ke ywords containing “CRISPR” as a substring, such as “CRISPR element metabolism” C1752766 , do appear . W e find this to be contradictory and that these inconsistencies highlight the limitation of relying on so strongly on k eyw ord databases. Going forward, we plan to de vise a way to extend a provided keyword network, utilizing semantic connections we can find within the MEDLINE document set. Projects lik e [42] hav e already shown this method can work in domains of smaller scales with good results. The challenge will be to extend this method to perform well when used on the entire MEDLINE data set. Impro ving Perf ormance of Algorithms with Graph Reordering T echniques: Cache-friendly layouts of graphs are known to generally accelerate the performance of the path and abstract retrie val algorithms which we apply . Moreov er , it is desirable to consider this type of acceleration in order to make our system more suitable for re gular modern desktops. This is an important consideration as memory is not expected to be a major bottleneck after the network is constructed. W e propose to rearrange the network nodes by minimizing such objectiv es as the minimum logarithmic or linear arrangements [36, 34]. On a mixture of K − K , A − K , and A − A edges we anticipate an improvement of at least 20% in the number of cache misses according to [35]. Mass Evaluation: W e note that ev aluation techniques are largely an issue in the state of the art of hypothesis generation. While some w orks feature large scale e valuation performed by man y human e xperts, a majority , this work included, are restricted to only a couple of promising results to jus tify the system. In order to better ev aluate and compare hypothesis generation techniques we must devise a common and large scale suite of historical hypotheses. W e are currently ev aluating whether a ground-truth network, like the drug- side-ef fect network SIDER [26], can be a good source of such hypotheses. For example, if we identify a set of recently added connections within SIDER, and predict a substantial percentage of those connections using MOLIERE , then we may be more certain of our performance. New Domains of Interest: W e ha ve considered other domains on which MOLIERE may perform well. These include generating hypotheses regarding economics, patents, narrative fiction, and social interactions. These are all domains where a hypothesis would in volve finding ne w relationships between distinct entities. W e contrast this with domains such as mathematics where the entity-relationship network is much less clear , and logical approaches from the field of automatic theorem proving are more applicable. 7 Conclusions In this study we describe a deployed biomedical hypothesis generation system, MOLIERE , that can dis- cov er relationship hypotheses among biomedical objects. This system utilizes information which exists in MEDLINE and other NLM datasets. W e validate MOLIERE on landmark discov eries using carefully fil- tered historical data. Unlike sev eral other hypothesis generation systems, we do not restrict the information retrie val domain to a specific language or a subset of scientific papers since this method can lose an unpre- dictable amount of information. Instead, we use recent text mining techniques that allo w us to work with the full heterogeneous data at scale. W e demonstrate that MOLIERE successfully generates hypotheses and recommend using it to advance biomedical kno wledge discovery . Going forward, we note a number of directions along which we can improv e MOLIERE as well as man y existing hypothesis generation systems. 16 8 Acknowledgments W e would like to thank Dr . Lihn Ngo for his help in using the Palmetto supercomputer which ran our experiments, and Cong Qiu for initial e xperiments with Neo4j. Refer ences [1] Specialist nlp tools, 2006. [2] Umls reference manual, 2009. [3] Pubmed, 2016. [4] Malorye Allison. Ncats launches drug repurposing program, 2012. [5] Christos Andronis, Anuj Sharma, V assilis V irvilis, Spyros Deftereos, and Aris Persidis. Literature mining, ontologies and information visualization for drug repurposing. Briefings in bioinformatics , 12(4):357–368, 2011. [6] Fredrik Barrenas, Sreeniv as Chav ali, Petter Holme, Reza Mobini, and Mikael Benson. Network prop- erties of comple x human disease genes ide ntified through genome-wide association studies. PloS one , 4(11):e8090, 2009. [7] David M Blei and John D Laf ferty . Dynamic topic models. In Pr oceedings of the 23r d international confer ence on Machine learning , pages 113–120. A CM, 2006. [8] David M Blei, Andrew Y Ng, and Michael I Jordan. Latent dirichlet allocation. J ournal of machine Learning r esearc h , 3(Jan):993–1022, 2003. [9] Piotr Bojanowski, Edouard Grav e, Armand Joulin, and T omas Mikolov . Enriching word vectors with subword information. , 2016. [10] Guus M Bol, F arhad V esuna, Min Xie, Jing Zeng, Khaled Aziz, Nishant Gandhi, Anne Le vine, Ashle y Irving, Dorian K orz, Saritha T antravedi, et al. T argeting ddx3 with a small molecule inhibitor for lung cancer therapy . EMBO molecular medicine , 7(5):648–669, 2015. [11] Guus Martinus Bol, Min Xie, and V enu Raman. Ddx3, a potential target for cancer treatment. Molec- ular cancer , 14(1):188, 2015. [12] HH Chen, HI Y u, WC Cho, and WY T arn. Ddx3 modulates cell adhesion and motility and cancer cell metastasis via rac1-mediated signaling pathway . Oncogene , 34(21):2790–2800, 2015. [13] Cristina-Maria Cruciat, Christine Dolde, Reinoud EA de Groot, Bisei Ohkaw ara, Carmen Reinhard, Hendrik C Korsw agen, and Christof Niehrs. Rna helicase ddx3 is a re gulatory sub unit of casein kinase 1 in wnt– β -catenin signaling. Science , 339(6126):1436–1441, 2013. [14] Neo4J Dev elopers. Neo4j. Graph NoSQL Database [online] , 2012. [15] Anna Di voli, Eneida A Mendonc ¸ a, James A Ev ans, and Andrey Rzhetsky . Conflicting biomed- ical assumptions for mathematical modeling: the case of cancer metastasis. PLoS Comput Biol , 7(10):e1002132, 2011. 17 [16] Ahmed El-Kishky , Y anglei Song, Chi W ang, Clare R V oss, and Jiawei Han. Scalable topical phrase mining from text corpora. Pr oceedings of the VLDB Endowment , 8(3):305–316, 2014. [17] James A Ev ans and Andre y Rzhetsky . Advancing science through mining libraries, ontologies, and communities. J ournal of Biological Chemistry , 286(27):23659–23666, 2011. [18] Jacob G Foster , Andrey Rzhetsky , and James A Evans. Tradition and innov ation in scientists research strategies. American Sociolo gical Revie w , 80(5):875–908, 2015. [19] Kwang-Il Goh, Michael E Cusick, David V alle, Barton Childs, Marc V idal, and Albert-L ´ aszl ´ o Barab ´ asi. The human disease network. Pr oceedings of the National Academy of Sciences , 104(21):8685–8690, 2007. [20] Thomas L Griffiths and Mark Steyvers. Finding scientific topics. Pr oceedings of the National academy of Sciences , 101(suppl 1):5228–5235, 2004. [21] Chris Gropp, Alexander Herzog, Ilya Safro, Paul W W ilson, and Amy W Apon. Scalable dynamic topic modeling with clustered latent dirichlet allocation (clda). , 2016. [22] Lei Hao, Zhongmin Zou, Hong T ian, Y ubo Zhang, Huchuan Zhou, and Lei Liu. Stem cell-based therapies for ischemic stroke. BioMed r esear ch international , 2014:468748, 2014. [23] Armand Joulin, Edouard Gra ve, Piotr Bojano wski, and T omas Mikolov . Bag of tricks for ef ficient text classification. , 2016. [24] Jacek Krol, Ilona Krol, Claudia P atricia Patino Alv arez, Michele Fiscella, Andreas Hierlemann, Botond Roska, and W itold Filipowicz. A network comprising short and long noncoding rnas and rna helicase controls mouse retina architecture. Natur e communications , 6, 2015. [25] Ann D Kwong, B Govinda Rao, and Kuan-T eh Jeang. V iral and cellular rna helicases as anti viral targets. Natur e r evie ws Drug discovery , 4(10):845–853, 2005. [26] Sejoon Lee, Kw ang H Lee, Mi n Song, and Doheon Lee. Building the process-drug–side effect net- work to discover the relationship between biological processes and side effects. BMC bioinformatics , 12(2):S2, 2011. [27] Chun-Chi Liu, Y u-T ing Tseng, W enyuan Li, Chia-Y u W u, Ilya Mayzus, Andre y Rzhetsky , Fengzhu Sun, Michael W aterman, Jeremy JW Chen, Preet M Chaudhary , et al. Diseaseconnect: a com- prehensi ve web server for mechanism-based disease–disease connections. Nucleic acids r esear ch , 42(W1):W137–W146, 2014. [28] Zhiyuan Liu, Y uzhou Zhang, Edward Y Chang, and Maosong Sun. Plda+: Parallel latent dirichlet allocation with data placement and pipeline processing. A CM T ransactions on Intelligent Systems and T echnology (TIST) , 2(3):26, 2011. [29] Giov anni Maga, Federico Falchi, Anna Garbelli, Amalia Belfiore, Myriam W itvrouw , Fabrizio Manetti, and Maurizio Botta. Pharmacophore modeling and molecular docking led to the disco very of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid- glutamic acid- alanine- aspartic acid box polypeptide 3. J ournal of medicinal chemistry , 51(21):6635– 6638, 2008. 18 [30] T omas Mikolov , Kai Chen, Greg Corrado, and Jeffre y Dean. Ef ficient estimation of word representa- tions in vector space. , 2013. [31] T omas Mikolo v , Ilya Sutske ver , Kai Chen, Greg S Corrado, and Jef f Dean. Distributed representa- tions of words and phrases and their compositionality . In Advances in neur al information pr ocessing systems , pages 3111–3119, 2013. [32] Marius Muja and Da vid G Lowe. Scalable nearest neighbor algorithms for high dimensional data. IEEE T ransactions on P attern Analysis and Machine Intelligence , 36(11):2227–2240, 2014. [33] TI Oprea and J Mestres. Drug repurposing: far beyond new targets for old drugs. The AAPS journal , 14(4):759–763, 2012. [34] I. Safro, D. Ron, and A. Brandt. Graph minimum linear arrangement by multile vel weighted edge contractions. J ournal of Algorithms , 60(1):24–41, 2006. [35] Ilya Safro, Paul D Ho vland, Jae wook Shin, and Michelle Mills Strout. Improving random walk per- formance. In CSC , pages 108–112, 2009. [36] Ilya Safro and Boris T emkin. Multiscale approach for the network compression-friendly ordering. J. Discr ete Algorithms , 9(2):190–202, 2011. [37] Sabindra K Samal, Samapika Routray , Ganesh Kumar V eeramachaneni, Rupesh Dash, and Mahendran Botlagunta. Ketorolac salt is a newly discov ered ddx3 inhibitor to treat oral cancer . Scientific r eports , 5:9982, 2015. [38] Neil R Smalheiser and Don R Sw anson. Using arro wsmith: a computer -assisted approach to formulat- ing and assessing scientific hypotheses. Computer methods and pr ograms in biomedicine , 57(3):149– 153, 1998. [39] Neil R Smalheiser , V etle I T orvik, and W ei Zhou. Arro wsmith two-node search interface: A tutorial on finding meaningful links between two disparate sets of articles in medline. Computer methods and pr ograms in biomedicine , 94(2):190–197, 2009. [40] Larisa N Soldatov a and Andrey Rzhetsk y . Representation of research hypotheses. Journal of biomed- ical semantics , 2(2):S9, 2011. [41] Scott Spangler . Accelerating Discovery: Mining Unstructur ed Information for Hypothesis Generation , volume 37. CRC Press, 2015. [42] Scott Spangler , Angela D W ilkins, Benjamin J Bachman, Meena Nagarajan, T ajhal Dayaram, Peter Haas, Sam Regenbogen, Curtis R Pickering, Austin Comer , Jef frey N Myers, et al. Automated h y- pothesis generation based on mining scientific literature. In Pr oceedings of the 20th A CM SIGKDD international confer ence on Knowledge discovery and data mining , pages 1877–1886. A CM, 2014. [43] Mianen Sun, Ling Song, T ong Zhou, G Y ancey Gillespie, and Richard S Jope. The role of ddx3 in regulating snail. Biochimica et Biophysica Acta (BBA)-Molecular Cell Resear ch , 1813(3):438–447, 2011. 19 [44] Don Swanson and Neil Smalheiser . Link analysis of medline titles as an aid to scientific disco very . In Pr oceedings of the AAAI F all Symposium on Artificial Intelligence and Link Analysis , pages 94–97, 1998. [45] Don R Swanson. Undiscov ered public kno wledge. The Library Quarterly , 56(2):103–118, 1986. [46] Don R Swanson and Neil R Smalheiser . Implicit text linkage between medline records: using arrow- smith as an aid to scientific discov ery . Library tr ends , 48(1):48, 1999. [47] Justin Sybrandt and Ilya Safro. Moliere: Automatic biomedical hypothesis generation system. medline kno wledge graph. implementation, 2017. [48] Marise R Heerma van V oss, W illemijne AME Schrijver , Natalie D ter Hoe ve, Laurien D Hoefnagel, Quirine F Manson, Elsken v an der W all, V enu Raman, P aul J van Diest, Dutch Distant Breast Can- cer Metastases Consortium, et al. The prognostic ef fect of ddx3 upre gulation in distant breast cancer metastases. Clinical & Experimental Metastasis , pages 1–8, 2016. [49] Huijun W ang, Y ing Ding, Jie T ang, Xiao Dong, Bing He, Judy Qiu, and David J W ild. Finding complex biological relationships in recent pubmed articles using bio-lda. PloS one , 6(3):e17243, 2011. [50] Daniel GR Y im and David M V irshup. Unwinding the wnt action of casein kinase 1. Cell r esearc h , 23(6):737, 2013. [51] XueZhong Zhou, J ¨ org Menche, Albert-L ´ aszl ´ o Barab ´ asi, and Amitabh Sharma. Human symptoms– disease network. Natur e communications , 5, 2014. 20
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment