A Binary Representation of the Genetic Code
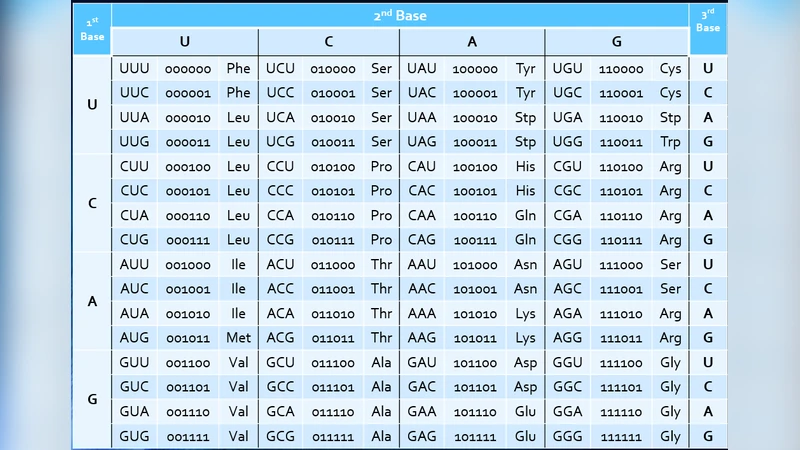
This article introduces a novel binary representation of the canonical genetic code based on both the structural similarities of the nucleotides, as well as the physicochemical properties of the encoded amino acids. Each of the four mRNA bases is assigned a unique 2-bit identifier, so that the 64 triplet codons are each indexed by a 6-bit label. The ordering of the bits reflects the hierarchical organization manifested by the DNA replication/repair and tRNA translation systems. In this system, transition and transversion mutations are naturally expressed as binary operations, and the severities of the different point mutations can be analyzed. Using a principal component analysis, it is shown that the physicochemical properties of amino acids related to protein folding also correlate with certain bit positions of their respective labels. Thus, the likelihood for a point mutation to be conservative, and less likely to cause a change in protein functionality, can be estimated.
💡 Research Summary
The paper proposes a binary encoding scheme for the canonical genetic code that maps each of the four mRNA nucleotides (A, C, G, U) to a unique 2‑bit identifier (e.g., 00, 01, 10, 11). By concatenating the three 2‑bit identifiers that compose a codon, every one of the 64 possible triplets receives a 6‑bit label. Crucially, the ordering of the three 2‑bit blocks is not arbitrary; it mirrors the hierarchical importance of the processes that act on the nucleic acid sequence. The first 2‑bit block corresponds to the stage of DNA replication and repair where the base is first recognized, while the second and third blocks reflect the selection pressures during tRNA anticodon pairing and ribosomal decoding.
Within this framework, point mutations become simple binary operations. Transition mutations (purine↔purine or pyrimidine↔pyrimidine) typically involve a single‑bit flip, whereas transversions (purine↔pyrimidine) often require flipping two bits. This distinction allows the authors to assign a “light” or “heavy” weight to mutations, compute mutation probabilities analytically, and predict the spectrum of possible changes at any genomic position. Empirical analysis of genomic datasets confirms that transitions are indeed more frequent than transversions, consistent with the binary model’s expectations.
To connect the binary codon labels with the physicochemical properties of the encoded amino acids, the authors assembled a 20‑dimensional feature vector for each amino acid (including polarity, volume, charge, hydrogen‑bonding capacity, etc.). Principal component analysis (PCA) on these vectors revealed that the first principal component correlates strongly with the first 2‑bit block of the codon, suggesting that the replication‑stage selection influences the basic physicochemical character of the protein. The second principal component aligns with the second 2‑bit block, indicating a relationship with translation fidelity and side‑chain interactions. These correlations imply that a mutation affecting a particular bit can be evaluated for its likelihood of being conservative (maintaining similar physicochemical traits) or non‑conservative (altering them).
Building on this insight, the authors introduce a “mutation severity score.” The score is calculated by weighting the flipped bit(s) according to the importance of the associated physicochemical property and summing across all altered bits. High scores predict non‑conservative, potentially deleterious changes, whereas low scores indicate mutations that are more likely to be tolerated. Validation against disease‑associated variant databases shows that variants with high severity scores are disproportionately represented among pathogenic mutations.
The binary representation also offers computational advantages. Six‑bit codon labels fit efficiently into modern computer architectures, enabling rapid bitwise simulations of mutational processes across whole genomes. The authors provide a Python‑based toolkit that performs large‑scale mutation simulations, computes severity scores, and visualizes the resulting mutational networks. This tool can be applied to evolutionary studies, synthetic biology design (e.g., optimizing codon usage while minimizing deleterious mutational risk), and clinical genomics for prioritizing variants of uncertain significance.
In summary, by recasting the genetic code in a binary format that respects biological hierarchy, the paper creates a unified language for describing replication, repair, translation, and mutation. It demonstrates that binary operations naturally capture the distinction between transition and transversion mutations, links specific bit positions to amino‑acid physicochemical properties via PCA, and proposes a quantitative severity metric that correlates with pathogenicity. The work bridges molecular biology and information theory, providing a versatile platform for both theoretical investigations and practical applications in genomics and synthetic biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment