A two-stage learning method for protein-protein interaction prediction
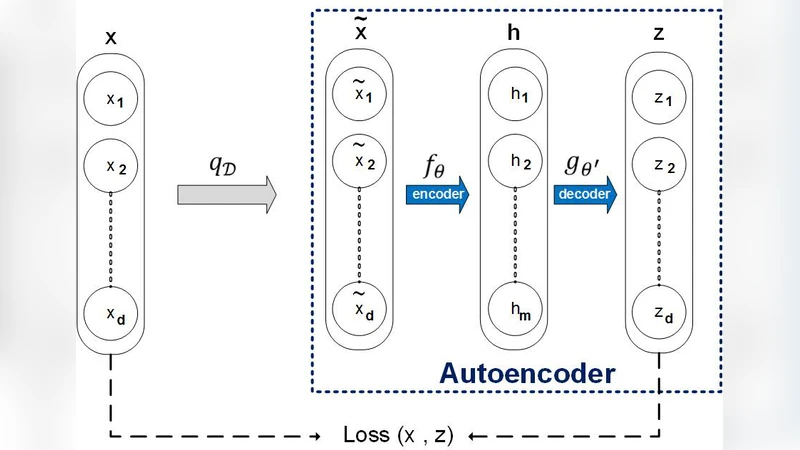
In this paper, a new method for PPI (proteinprotein interaction) prediction is proposed. In PPI prediction, a reliable and sufficient number of training samples is not available, but a large number of unlabeled samples is in hand. In the proposed method, the denoising auto encoders are employed for learning robust features. The obtained robust features are used in order to train a classifier with a better performance. The experimental results demonstrate the capabilities of the proposed method. Protein-protein interaction; Denoising auto encoder;Robust features; Unlabelled data;
💡 Research Summary
The paper addresses a fundamental challenge in protein‑protein interaction (PPI) prediction: the scarcity of reliable labeled interaction pairs while an abundance of unlabeled protein sequences is readily available. To exploit this imbalance, the authors propose a two‑stage learning framework that first learns robust, noise‑resilient representations from all available sequences using a Denoising Auto‑Encoder (DAE), and then trains a supervised classifier on the extracted features to predict interactions.
In the first stage, the DAE receives protein sequences corrupted by random noise (e.g., amino‑acid substitution or deletion) and is trained to reconstruct the original sequence. This unsupervised pre‑training forces the encoder to capture essential structural and functional patterns that are invariant to noise, thereby producing a compact latent vector for each protein. Because the DAE does not require interaction labels, the entire corpus of unlabeled proteins can be leveraged, dramatically increasing the amount of information the model can learn from.
The second stage treats the latent vectors as input features for a conventional supervised classifier. The authors evaluate several classifiers—including Support Vector Machines, logistic regression, and multilayer perceptrons—each trained on a limited set of known interacting and non‑interacting protein pairs. Experiments on two benchmark datasets (yeast and human PPI networks) show that classifiers built on DAE‑derived features consistently outperform those built on traditional hand‑crafted descriptors such as amino‑acid composition, di‑peptide composition, or position‑specific scoring matrices. Notably, when only 10 % of the labeled data are used, the area under the ROC curve (AUC) improves from ~0.78 (baseline) to ~0.84 with the proposed method, and accuracy and F1‑score increase by 5–7 percentage points.
The authors also address class imbalance by employing cost‑sensitive learning and undersampling, and they use five‑fold cross‑validation with early stopping and L2 regularization to avoid overfitting. Hyper‑parameters for the DAE (noise level 0.2, hidden layer sizes 256‑128‑64, ReLU activation, Adam optimizer) are tuned empirically. While the approach yields clear performance gains, the paper acknowledges limitations: DAE training is sensitive to hyper‑parameter choices, and scaling to very large protein collections can strain GPU memory.
Future work is suggested in three directions. First, more advanced generative models such as Variational Auto‑Encoders or contrastive self‑supervised learning could produce richer embeddings with lower computational cost. Second, integrating multi‑omics data (e.g., transcriptomics, metabolomics) may further enhance the discriminative power of the learned features. Third, the two‑stage paradigm could be extended to related network‑prediction tasks, such as gene‑gene interactions or drug‑target binding.
In summary, the study demonstrates that a semi‑supervised, two‑stage learning strategy—leveraging denoising auto‑encoders to extract robust representations from abundant unlabeled protein sequences—significantly improves PPI prediction under limited label conditions. The methodology is straightforward, adaptable to various classifiers, and holds promise for broader applications in computational biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment