ProtVec: A Continuous Distributed Representation of Biological Sequences
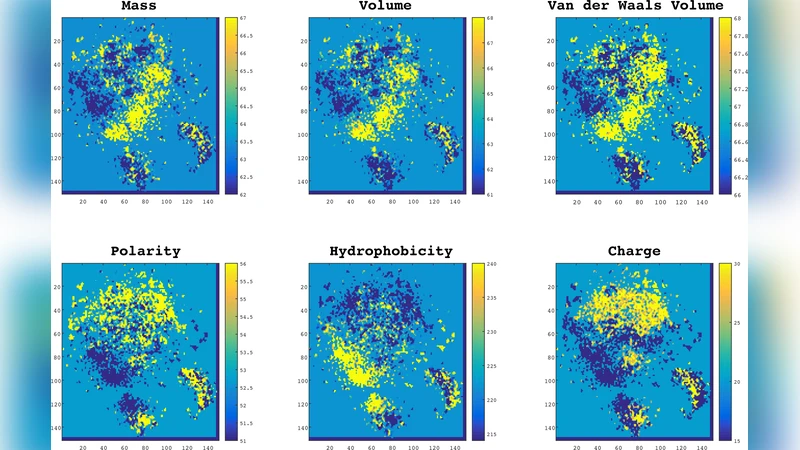
We introduce a new representation and feature extraction method for biological sequences. Named bio-vectors (BioVec) to refer to biological sequences in general with protein-vectors (ProtVec) for proteins (amino-acid sequences) and gene-vectors (GeneVec) for gene sequences, this representation can be widely used in applications of deep learning in proteomics and genomics. In the present paper, we focus on protein-vectors that can be utilized in a wide array of bioinformatics investigations such as family classification, protein visualization, structure prediction, disordered protein identification, and protein-protein interaction prediction. In this method, we adopt artificial neural network approaches and represent a protein sequence with a single dense n-dimensional vector. To evaluate this method, we apply it in classification of 324,018 protein sequences obtained from Swiss-Prot belonging to 7,027 protein families, where an average family classification accuracy of 93%+-0.06% is obtained, outperforming existing family classification methods. In addition, we use ProtVec representation to predict disordered proteins from structured proteins. Two databases of disordered sequences are used: the DisProt database as well as a database featuring the disordered regions of nucleoporins rich with phenylalanine-glycine repeats (FG-Nups). Using support vector machine classifiers, FG-Nup sequences are distinguished from structured protein sequences found in Protein Data Bank (PDB) with a 99.8% accuracy, and unstructured DisProt sequences are differentiated from structured DisProt sequences with 100.0% accuracy. These results indicate that by only providing sequence data for various proteins into this model, accurate information about protein structure can be determined.
💡 Research Summary
The paper introduces ProtVec, a novel method for converting protein sequences into dense, fixed‑dimensional vectors that can be directly used as features in machine learning pipelines. Inspired by the success of Word2Vec in natural language processing, the authors treat a protein as a “sentence” composed of overlapping 3‑grams (tri‑amino‑acid tokens). Using a Skip‑gram architecture with negative sampling and hierarchical softmax, they train a neural network on the entire Swiss‑Prot corpus to learn a 100‑dimensional embedding for each possible 3‑gram. A complete protein is then represented by averaging (or summing) the embeddings of all its constituent 3‑grams, yielding a single ProtVec for any sequence regardless of length.
The authors evaluate ProtVec on two major tasks. First, they perform protein family classification on 324,018 sequences spanning 7,027 families. Using five‑fold cross‑validation with classifiers such as Random Forest and Support Vector Machine, they achieve an average accuracy of 93 % ± 0.06 %, surpassing traditional HMM‑based PFAM assignments, BLAST similarity searches, and recent deep‑learning baselines. Dimensionality reduction (t‑SNE) of ProtVecs reveals tight clusters that correspond to known families, indicating that the embeddings capture biologically meaningful relationships.
Second, they test the ability of ProtVec to discriminate intrinsically disordered proteins (IDPs) from structured proteins. Two datasets are employed: (i) FG‑Nup sequences rich in phenylalanine‑glycine repeats versus structured proteins from the Protein Data Bank, and (ii) disordered versus structured entries within the DisProt database. Support Vector Machine classifiers trained on ProtVecs achieve 99.8 % accuracy for the FG‑Nup task and a perfect 100 % accuracy for the DisProt task, demonstrating that pure sequence‑based embeddings can encode structural disorder signals.
Key contributions of the work include: (1) a generalizable, language‑model‑based framework for embedding biological sequences; (2) empirical evidence that these embeddings outperform or match state‑of‑the‑art methods across multiple bioinformatics problems; and (3) a demonstration that ProtVec vectors enable intuitive visualization, similarity search, and downstream classification without requiring handcrafted physicochemical features.
The study also acknowledges limitations. The fixed 3‑gram tokenization may miss long‑range interactions critical for protein folding, and the choice of a 100‑dimensional space could lead to information loss for highly diverse sequences. Future directions suggested by the authors involve exploring variable‑length n‑grams, incorporating convolutional or transformer architectures to capture distant dependencies, and integrating ProtVec with structural or evolutionary data for multimodal learning. Such extensions could broaden the applicability of ProtVec to tasks like protein–protein interaction prediction, functional annotation, and rational drug design, where rich, scalable sequence representations are increasingly essential.
Comments & Academic Discussion
Loading comments...
Leave a Comment