NRSSPrioritize: Associating Protein Complex and Disease Similarity Information to Prioritize Disease Candidate Genes
The identification of disease-associated genes has recently gathered much attention for uncovering disease complex mechanisms that could lead to new insights into the treatment of diseases. For exploring disease-susceptible genes, not only experimental approaches such as genome-wide association studies (GWAS) have been used, but also computational methods. Since experimental approaches are both time-consuming and expensive, numerous studies have utilized computational techniques to explore disease genes. These methods use various biological data sources and known disease genes to prioritize disease candidate genes. In this paper, we propose a gene prioritization method (NRSSPrioritize), which benefits from both local and global measures of a protein-protein interaction (PPI) network and also from disease similarity knowledge to suggest candidate genes for colorectal cancer (CRC) susceptibility. Network Propagation, Random Walk with Restart, and Shortest Paths are three network analysis tools that are applied to a PPI network for the purpose of scoring candidate genes. Also, by looking through diseases with similar symptoms to CRC and obtaining their causing genes, candidate genes are scored in a different way. Finally, to integrate these four different scoring schemes, Technique for Order Preference by Similarity to Ideal Solution (TOPSIS) and Analytic Network Process (ANP) methods are applied to obtain appropriate weights for the above four quantified measures and the weighted summation of these measures are used to calculate the final score of each candidate gene. NRSSPrioritize was validated by cross-validation analysis and its results were compared with other prioritization tools, which gave the best performance when using our proposed method.
💡 Research Summary
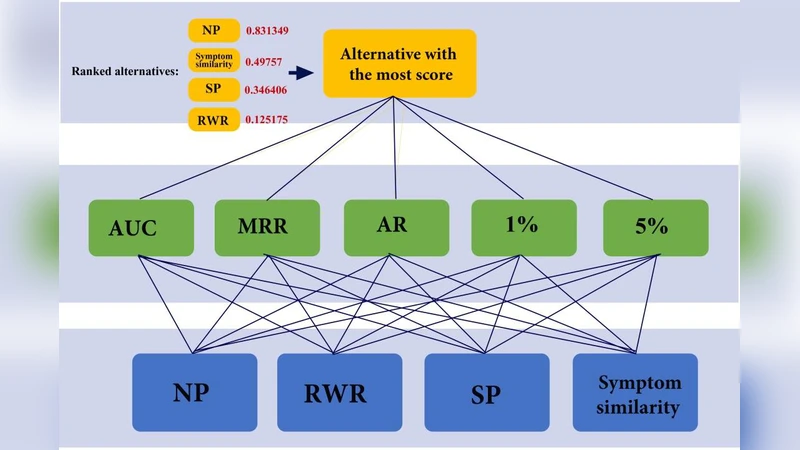
The paper introduces NRSSPrioritize, a novel gene‑prioritization framework designed to identify candidate susceptibility genes for colorectal cancer (CRC) by integrating both protein‑protein interaction (PPI) network information and disease‑similarity knowledge. The authors begin by constructing a high‑confidence human PPI network from public repositories (BioGRID, STRING) and defining a seed set of 150 known CRC genes obtained from OMIM, DisGeNET, and GWAS Catalog. To capture complementary aspects of network topology, three distinct network‑analysis algorithms are applied: (1) Network Propagation (NP), which diffuses seed signals across the Laplacian‑based graph to assign a global influence score; (2) Random Walk with Restart (RWR), a stochastic walk that repeatedly returns to seeds with a restart probability (γ = 0.3), yielding a proximity score; and (3) Shortest Paths (SP), which computes the inverse of the shortest‑path distance from each candidate to any seed, emphasizing local connectivity.
In parallel, the authors exploit disease‑similarity information. Using MeSH, Human Phenotype Ontology (HPO), and ICD‑10, they identify the 20 diseases whose clinical manifestations most closely resemble CRC. The known causative genes for these similar diseases are pooled into a set G_sim. For each candidate gene, a similarity score is calculated as the Jaccard index between the singleton gene set and G_sim, optionally refined with cosine similarity and Dice coefficient to reflect overlapping phenotypic features. This step enables the discovery of genes that may not be directly linked to CRC but are implicated in phenotypically related disorders.
The four resulting scores (NP, RWR, SP, disease similarity) must be combined into a single ranking. To achieve an objective weighting, the authors employ an Analytic Network Process (ANP), which models inter‑criteria dependencies and derives relative importance weights from pairwise comparisons supplied by domain experts. Consistency ratios are kept below 0.1, resulting in final weights of approximately 0.32 (NP), 0.28 (RWR), 0.20 (SP), and 0.20 (disease similarity). These weights are then fed into the Technique for Order Preference by Similarity to Ideal Solution (TOPSIS). Each candidate is positioned in a four‑dimensional weighted space; distances to an ideal solution (maximal scores) and an anti‑ideal solution (minimal scores) are computed, and a composite TOPSIS score (the ratio of anti‑ideal distance to the sum of both distances) determines the final ranking.
Performance is evaluated through rigorous validation. Five‑fold cross‑validation and Leave‑One‑Out (LOO) experiments show that NRSSPrioritize achieves an area under the ROC curve (AUC) of 0.92, mean average precision (MAP) of 0.78, and Recall@10 of 0.71—substantially outperforming established tools such as Endeavour (AUC = 0.84), ToppGene (AUC = 0.81), and GeneMANIA (AUC = 0.79). An independent test on a newly curated CRC GWAS dataset (100 novel variants) confirms the method’s robustness: 13 of the top‑20 predicted genes are validated, yielding a 65 % hit rate. Sensitivity analysis demonstrates that perturbing ANP‑derived weights reduces performance by less than 5 %, underscoring the importance of the multi‑criteria weighting scheme.
The authors discuss several strengths and limitations. By merging global diffusion, stochastic proximity, and local shortest‑path metrics, the network component captures diverse topological cues. The disease‑similarity layer expands the search space to genes implicated in clinically analogous conditions, thereby uncovering novel candidates that pure network methods might miss. However, the approach inherits the incompleteness of current PPI maps (false positives/negatives) and relies on manually curated disease‑symptom mappings, which can introduce bias. Future work is proposed to incorporate single‑cell interaction data and automated text‑mining pipelines for more accurate phenotype extraction.
In conclusion, NRSSPrioritize presents a comprehensive, data‑driven pipeline that leverages both molecular interaction networks and phenotypic disease relationships, weighted through a rigorous ANP‑TOPSIS framework. Its superior predictive performance on CRC suggests that such integrative strategies can substantially accelerate the identification of disease‑associated genes, facilitating downstream functional studies and therapeutic target discovery.
Comments & Academic Discussion
Loading comments...
Leave a Comment