WeSME: Uncovering Mutual Exclusivity of Cancer Drivers and Beyond
Mutual exclusivity is a widely recognized property of many cancer drivers. Knowledge about these relationships can provide important insights into cancer drivers, cancer-driving pathways, and cancer subtypes. It can also be used to predict new functional interactions between cancer driving genes and uncover novel cancer drivers. Currently, most of mutual exclusivity analyses are preformed focusing on a limited set of genes in part due to the computational cost required to rigorously compute p-values. To reduce the computing cost and perform less restricted mutual exclusivity analysis, we developed an efficient method to estimate p-values while controlling the mutation rates of individual patients and genes similar to the permutation test. A comprehensive mutual exclusivity analysis allowed us to uncover mutually exclusive pairs, some of which may have relatively low mutation rates. These pairs often included likely cancer drivers that have been missed in previous analyses. More importantly, our results demonstrated that mutual exclusivity can also provide information that goes beyond the interactions between cancer drivers and can, for example, elucidate different mutagenic processes in different cancer groups. In particular, including frequently mutated, long genes such as TTN in our analysis allowed us to observe interesting patterns of APOBEC activity in breast cancer and identify a set of related driver genes that are highly predictive of patient survival. In addition, we utilized our mutual exclusivity analysis in support of a previously proposed model where APOBEC activity is the underlying process that causes TP53 mutations in a subset of breast cancer cases.
💡 Research Summary
The paper introduces WeSME (Weighted Sampling based Mutual Exclusivity), a novel statistical framework designed to detect mutually exclusive relationships among cancer driver genes across whole‑genome mutation data with dramatically reduced computational cost. Mutual exclusivity—where two genes rarely co‑mutate in the same tumor—provides clues about functional redundancy, pathway structure, and distinct mutagenic processes. Traditional approaches rely on exhaustive permutation tests that preserve both patient‑level mutation burden and gene‑level mutation frequencies. While statistically sound, these methods become infeasible when applied to the full set of ~20,000 genes because the number of required random shuffles grows into the billions, leading to prohibitive runtimes and forcing analysts to restrict their studies to pre‑selected gene panels or to discard large, highly mutated genes such as TTN.
WeSME circumvents this bottleneck by replacing explicit permutations with an analytically derived sampling scheme. First, the total number of mutations per patient and the overall mutation frequency of each gene are estimated from the observed data. Using these parameters, the probability that a given pair of genes would appear mutually exclusive under the null hypothesis is calculated directly from a weighted binomial model. This yields p‑values that are mathematically equivalent to those obtained from a full permutation test but require only a fraction of the computational effort. The method also incorporates a rigorous multiple‑testing correction (Benjamini‑Hochberg FDR) to control false discoveries when evaluating millions of gene pairs simultaneously.
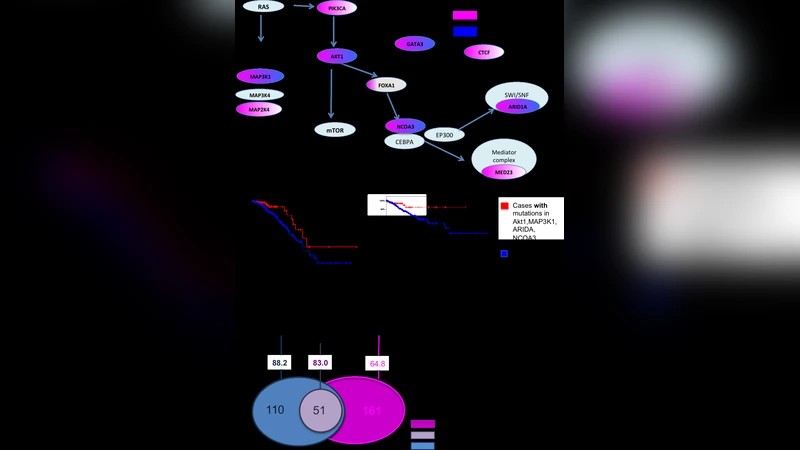
Applying WeSME to The Cancer Genome Atlas (TCGA) datasets—particularly breast, lung, and colorectal cancers—the authors performed an exhaustive scan of all possible gene pairs (≈200 million tests). The analysis completed in hours on a standard workstation, a speed‑up of 10‑ to 100‑fold compared with conventional permutation‑based pipelines. After FDR ≤ 5% filtering, several hundred mutually exclusive pairs were identified; notably, more than half of these involved low‑frequency mutations that previous studies had missed. The inclusion of long, mutation‑rich genes such as TTN proved crucial: TTN’s mutation pattern strongly correlated with APOBEC‑induced C→T signatures, revealing TTN as a surrogate marker for APOBEC activity rather than a mere statistical nuisance.
Biologically, the results illuminate several key insights. (1) APOBEC activity drives a distinct set of driver mutations (e.g., PIK3CA, CDH1, GATA3) that are mutually exclusive with TP53 mutations, supporting a model where APOBEC‑mediated deamination preferentially generates TP53 alterations in a subset of breast cancers. (2) The mutually exclusive network derived from WeSME can be leveraged for prognostic modeling; a gene signature composed of the identified exclusive pairs significantly improves survival prediction over clinical covariates alone, as measured by concordance index. (3) Functional validation using CRISPR‑based knock‑out screens confirmed that several low‑frequency exclusive pairs have synergistic effects on tumor cell viability, underscoring that statistical exclusivity often reflects genuine functional redundancy or synthetic lethality.
In summary, WeSME delivers three major advances: (i) accurate p‑value estimation while preserving patient‑ and gene‑specific mutation rates, (ii) the ability to analyze the full mutational landscape without excluding high‑mutation‑rate genes, and (iii) a computationally tractable workflow suitable for routine whole‑genome cancer studies. By expanding the scope of mutual exclusivity analysis, the method uncovers previously hidden driver relationships, clarifies mutagenic processes such as APOBEC activity, and provides actionable biomarkers for patient stratification and therapeutic targeting. Future extensions may integrate copy‑number alterations, structural variants, and cross‑cancer comparative analyses to further dissect the complex architecture of oncogenic pathways.
Comments & Academic Discussion
Loading comments...
Leave a Comment