Computational characterization and prediction of metal-organic framework properties
In this introductory review, we give an overview of the computational chemistry methods commonly used in the field of metal-organic frameworks (MOFs), to describe or predict the structures themselves and characterize their various properties, either at the quantum chemical level or through classical molecular simulation. We discuss the methods for the prediction of crystal structures, geometrical properties and large-scale screening of hypothetical MOFs, as well as their thermal and mechanical properties. A separate section deals with the simulation of adsorption of fluids and fluid mixtures in MOFs.
💡 Research Summary
The review by Coudert and Fuchs provides a comprehensive overview of computational methods used to describe, predict, and characterize metal‑organic frameworks (MOFs) at both the quantum‑chemical and classical simulation levels. It begins with an introduction that situates molecular simulation within the broader history of MOF research, emphasizing that techniques such as density‑functional theory (DFT) and Grand‑Canonical Monte Carlo (GCMC) have become routine tools accessible even on modest hardware.
The structural‑property section is divided into five subsections. The first discusses crystal‑structure prediction (CSP). Traditional CSP approaches—simulated annealing, genetic algorithms, and evolutionary strategies—are described, followed by the Automated Assembly of Secondary Building Units (AASBU) method, which uses predefined SBUs with “sticky‑atom” potentials to self‑assemble frameworks in silico. While AASBU can reproduce known structures (e.g., HKUST‑1, MOF‑5) and propose new ones, its high computational cost limits large‑scale use. The second subsection addresses large‑scale screening and the generation of hypothetical MOF databases. The authors highlight the “LEGO‑like” bottom‑up approach pioneered by Wilmer et al., where experimentally known metal nodes and organic linkers are combined according to geometric attachment rules, yielding on the order of 10⁵–10⁶ candidate structures without the need for prior energy minimization. The third subsection covers geometrical descriptors such as surface area, pore volume, and pore‑size distribution, typically computed with tools like Zeo++ using Connolly surfaces or Voronoi tessellations. The fourth subsection introduces advanced descriptors (fractal dimension, accessibility indices) that capture subtle aspects of pore topology and are useful for machine‑learning models. The fifth subsection discusses the localization of extra‑framework ions, which requires a combination of electrostatic calculations, DFT charge analysis, and classical force‑field simulations.
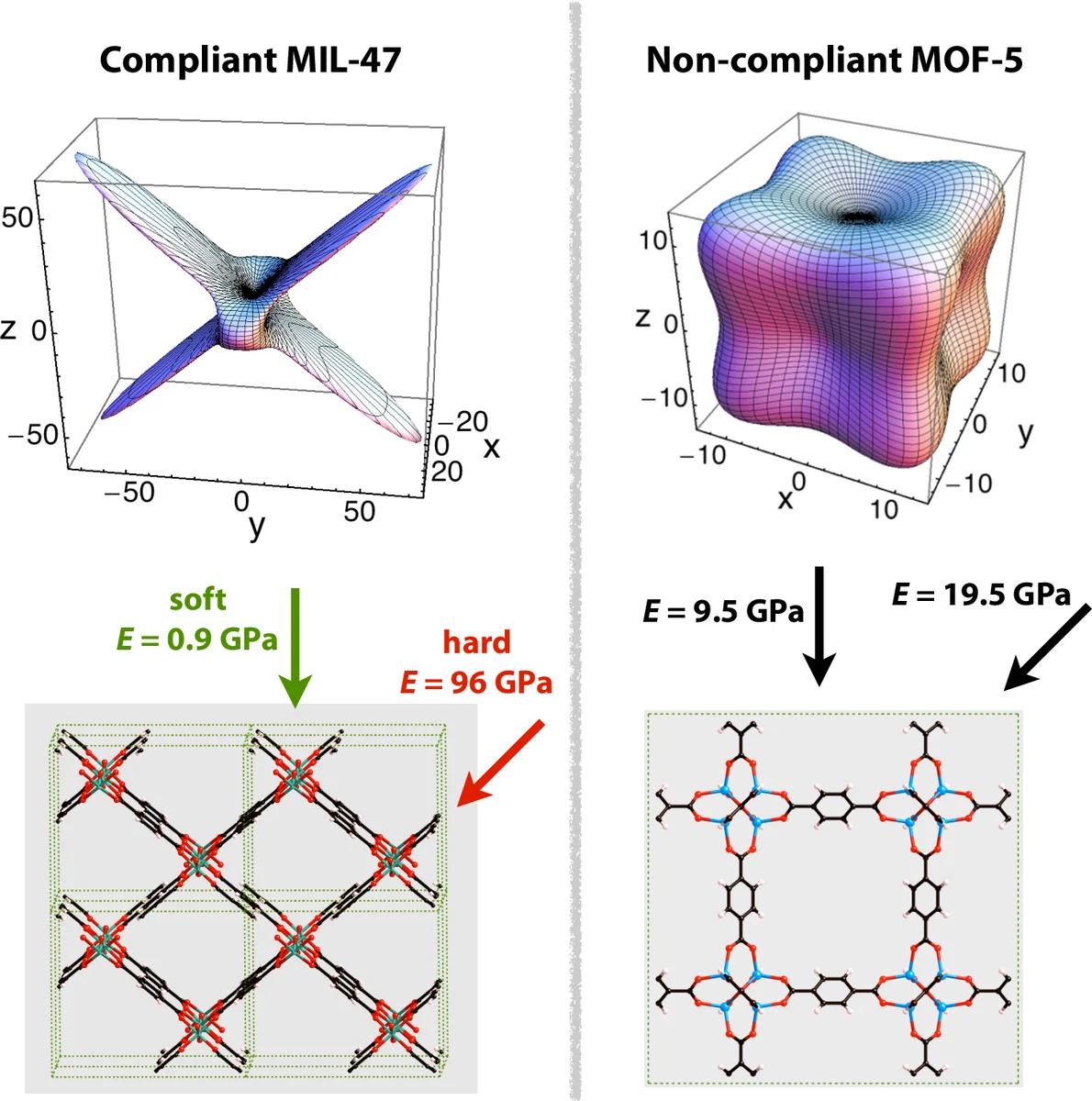
Physical properties are examined in three parts. Mechanical properties are obtained from elastic‑tensor calculations via DFT or from stress‑strain molecular‑dynamics simulations, providing bulk modulus, shear modulus, and Poisson’s ratio. Thermal properties—including thermal expansion coefficients, heat capacity, and lattice thermal conductivity—are derived from phonon calculations or from equilibrium/non‑equilibrium MD. Optical and electronic properties are addressed through band‑structure analysis, density‑of‑states calculations, and time‑dependent DFT to predict absorption spectra and potential semiconducting behavior.
The adsorption section is the most extensive. GCMC is presented as the workhorse for predicting single‑component gas uptake, with emphasis on the need for accurate force fields that capture Lennard‑Jones dispersion, electrostatics, and, when relevant, specific interactions at open metal sites. Multi‑component (co‑adsorption) simulations are discussed, along with methods for calculating selectivity and mixture isotherms. The authors note that classical force fields may fail for chemisorption; in such cases, hybrid quantum‑mechanical/molecular‑mechanical (QM/MM) or DFT‑based adsorption energy calculations are required. Flexible MOFs, whose frameworks deform upon adsorption, are treated with hybrid GCMC/MD or with the use of the osmotic ensemble, allowing the coupling of host deformation and guest loading. Finally, the authors stress the importance of systematic comparison with experimental data, including the use of consistent reference states, accounting for framework defects, and acknowledging uncertainties in both simulation and measurement.
The perspective part outlines three forward‑looking challenges. First, modeling defects and disorder remains difficult; stochastic approaches or explicit defect insertion in supercells are suggested. Second, the creation and curation of large, open‑access databases (e.g., CoRE‑MOF, QMOF) are essential for high‑throughput screening and for training data‑driven models. Third, stimuli‑responsive MOFs—those that change structure under pressure, temperature, light, or electric fields—require multi‑scale simulations that combine quantum chemistry, classical MD, and continuum models.
Overall, the review underscores that computational chemistry, when integrated with experimental validation, can dramatically accelerate MOF discovery, guide synthesis, and enable rational design of materials for gas storage, separation, catalysis, and beyond.
Comments & Academic Discussion
Loading comments...
Leave a Comment