Discovering functional DNA elements using population genomic information: A proof of concept using human mtDNA
Identifying the complete set of functional elements within the human genome would be a windfall for multiple areas of biological research including medicine, molecular biology, and evolution. Complete knowledge of function would aid in the prioritization of loci when searching for the genetic bases of disease or adaptive phenotypes. Because mutations that disrupt function are disfavored by natural selection, purifying selection leaves a detectable signature within functional elements; accordingly this signal has been exploited through the use of genomic comparisons of distantly related species. However, the functional complement of the genome changes extensively across time and between lineages, therefore, evidence of the current action of purifying selection is essential. Because the removal of deleterious mutations by natural selection also reduces within-species genetic diversity within functional loci, dense population genetic data have the potential to reveal genomic elements that are currently functional. Here we assess the potential of this approach using 16,411 human mitochondrial genomes. We show that the high density of polymorphism in this dataset precisely delineates regions experiencing purifying selection. Further, we show that the number of segregating alleles at a site is strongly correlated with its divergence across species after accounting for known mutational biases in human mtDNA. These two measures track one another at a remarkably fine scale across many loci–a correlation that is purely the result of natural selection. Our results demonstrate that genetic variation has the potential to reveal exactly which nucleotides in the genome are currently performing important functions and likely to have deleterious fitness effects when mutated. As more complete genomes are sequenced, similar power to reveal purifying selection may be achievable in the human nuclear genome.
💡 Research Summary
The paper presents a proof‑of‑concept that dense population‑genomic data can be used to map currently functional DNA elements by detecting the footprint of ongoing purifying selection. Traditional approaches rely on inter‑species conservation, which only captures elements that have been conserved over long evolutionary periods and may miss lineage‑specific or recently evolved functions. In contrast, deleterious mutations that are being removed by natural selection reduce within‑species polymorphism at functional sites, providing a contemporaneous signal of functional constraint.
The authors assembled a dataset of 16,411 complete human mitochondrial genomes (mtDNA), a system that is ideal for this analysis because of its high copy number, lack of recombination, and the availability of a very large number of sequences from diverse populations. For each nucleotide position they counted the number of segregating alleles (polymorphism density) and compared this landscape to known functional annotations (rRNA, tRNA, protein‑coding genes) and to measures of inter‑species divergence (human versus chimpanzee, mouse, whale, etc.).
Key findings:
- Regions with low polymorphism density coincide precisely with annotated functional elements. tRNA and rRNA genes, as well as conserved codon positions within protein‑coding genes, show markedly fewer segregating alleles than non‑coding intergenic segments. This pattern is consistent with strong purifying selection eliminating deleterious variants.
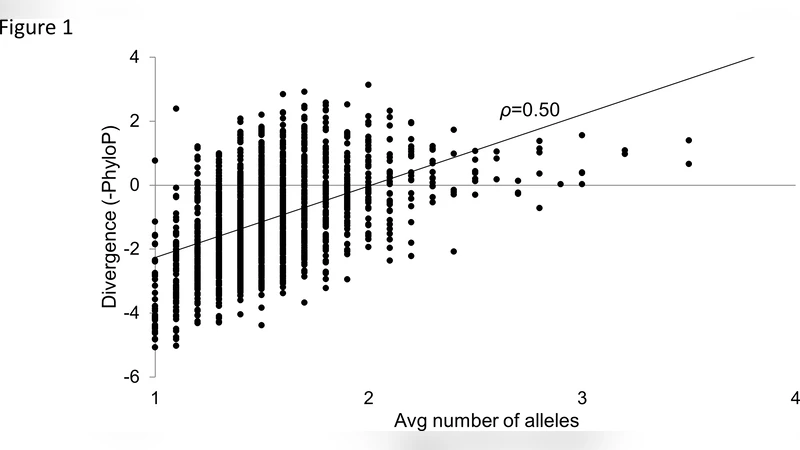
- Polymorphism density is positively correlated with nucleotide divergence across species. Sites that accumulate many alleles within humans also tend to be more divergent between species, while highly conserved sites exhibit both low within‑species variation and low between‑species divergence. The correlation persists after correcting for known mtDNA mutational biases (e.g., transition‑transversion asymmetry), indicating that the relationship is driven by selection rather than mutational hotspots.
- The correlation holds at a very fine scale—down to single‑nucleotide resolution—demonstrating that population variation can pinpoint functional constraint at the base‑pair level, a resolution far beyond that achievable with conventional comparative genomics, which typically identifies broader conserved blocks.
The authors discuss the broader implications. Because the signal derives from the removal of deleterious alleles, it reflects the current functional state of the genome, not just historical conservation. This makes the approach valuable for prioritizing variants in medical genetics: a nucleotide with low polymorphism in a large human cohort is likely under strong functional constraint and any mutation there may have a high probability of pathogenicity. Moreover, as whole‑genome sequencing projects continue to expand (e.g., gnomAD, TOPMed), the same methodology can be applied to the nuclear genome. However, the nuclear genome presents additional challenges—recombination, heterogeneous mutation rates, and structural variation—that will require more sophisticated statistical models and larger sample sizes to achieve comparable resolution.
In conclusion, the study validates that population‑genomic data can serve as a direct, high‑resolution map of purifying selection, accurately delineating functional DNA elements in the human mitochondrial genome. The success of this proof‑of‑concept suggests that, with sufficient sequencing depth, similar analyses will become a powerful tool for annotating functional regions across the entire human genome, improving our ability to interpret genetic variation in health and disease.